Explorer enables data scientists and researchers to focus on answering biological questions by abstracting away the need for managing scalable computational infrastructure.
Although Explorer is built with special consideration for genomics applications and Gencove datasets in mind, it is a general-purpose computing platform on which general scientific computing can be easily performed.
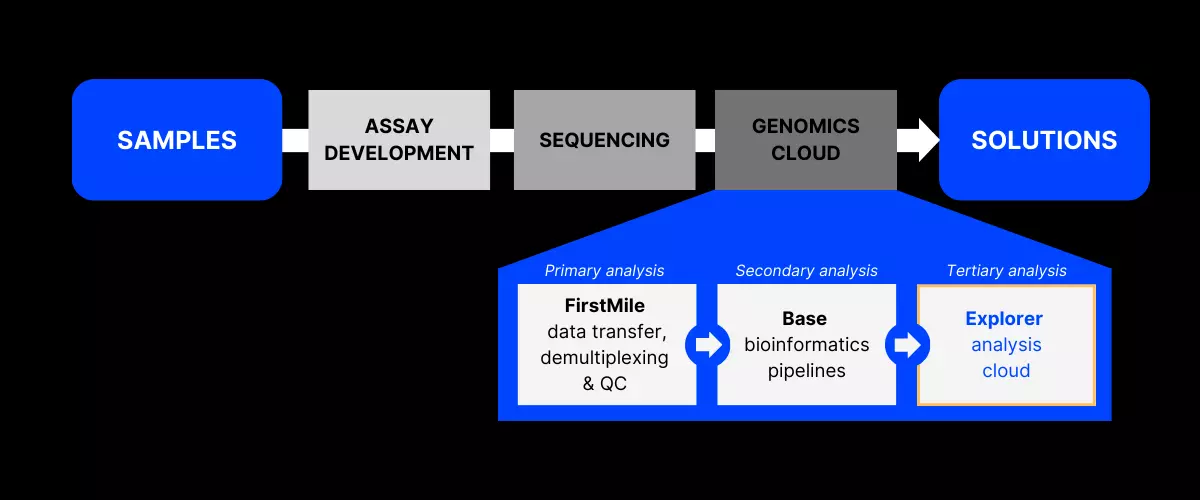
Explorer allows you to organize, analyze, and visualize your data all in one place. At a high level, Explorer comprises the following systems:
- An Explorer instance, which is a dedicated Linux machine with which you interact through a hosted JupyterLab instance
- A scalable compute cluster (up to tens of thousands of CPUs), to which you can submit arbitrary analysis jobs from within your Explorer instance
- A persistent object store providing private cloud storage, on which you can store your data and analysis outputs
- The Explorer SDK, a lightweight Python package which makes it easy to interface with both the compute cluster and object storage and which additionally provides a native integration with Gencove-generated data
- Explorer Shortcuts, a Python package which provides off-the-shelf functions to perform commonly required tasks such as merging VCFs, filtering alignments, VCF annotation, genotype concordance calculations, and more
For those with a more traditional cluster computing background, one can think of the Explorer instance as the head node of a cluster, on which light analyses can be performed and from which jobs that require heavy lifting can be submitted to the Batch cluster through use of the Explorer SDK. The difference here is that persistent object storage is achieved through the use of private cloud storage rather than, for example, a shared filesystem on the compute nodes of the cluster.
A variety of abstractions for cluster jobs and input/output files are implemented in the Explorer SDK.
Particularly of note is an abstraction of an "analysis" (implemented through a Python Analysis object), which encapsulates a "work function", a user-defined function which allows interleaved shell and Python code, along with the inputs and outputs associated with a job that is submitted to the Explorer cluster. This abstraction enables the straightforward definition of an arbitrary analysis that can leverage both bioinformatics shell tools (e.g., PLINK, samtools, bcftools, etc.) as well as Python.
Furthermore, the SDK enables the definition of dependencies between multiple different analysis jobs, such that entire workflows with various sequential and interdependent steps can easily be defined and executed on the Explorer-provided cluster (without having to manage the underlying infrastructure) as long as the workflow can be represented as a directed acyclic graph (DAG).
By bundling an on-demand computing cluster with a genomics-oriented SDK and native integration with other existing elements of the Gencove platform, data science and research teams are empowered to spend their time focusing on the analytical elements of scientific research rather than dealing with the dual challenges of managing complex infrastructure and the operational complexity that comes with having to navigate multiple systems.