Introduction
In this notebook we will demonstrate how to use Gencove Explorer to perform genomic prediction in maize.
Specifically, we will be building a genomic prediction model for maize grain yield. Genomic prediction is a technique from statistical genetics that uses genome-wide genetic markers to estimate the breeding value (sometimes referred to as genetic merit) of plants or animals in modern breeding programs. This estimated breeding value (EBV) can be used to identify and selectively breed the best individuals in the population in order to improve the agronomically relevant traits.
Genomic prediction has revolutionized breeding programs across various agricultural species due to its cost-effectiveness and accuracy. Genomic prediction often approaches the accuracy of direct phenotyping but at a fraction of the cost. Additionally, it enables the estimation of breeding values for traits not directly expressed by an individual, such as a dairy bull's genetic potential for milk quality traits in his female offspring. For an in-depth understanding of the goals and foundational theory of genomic prediction, see the original work in the field by Meuwissen et al.
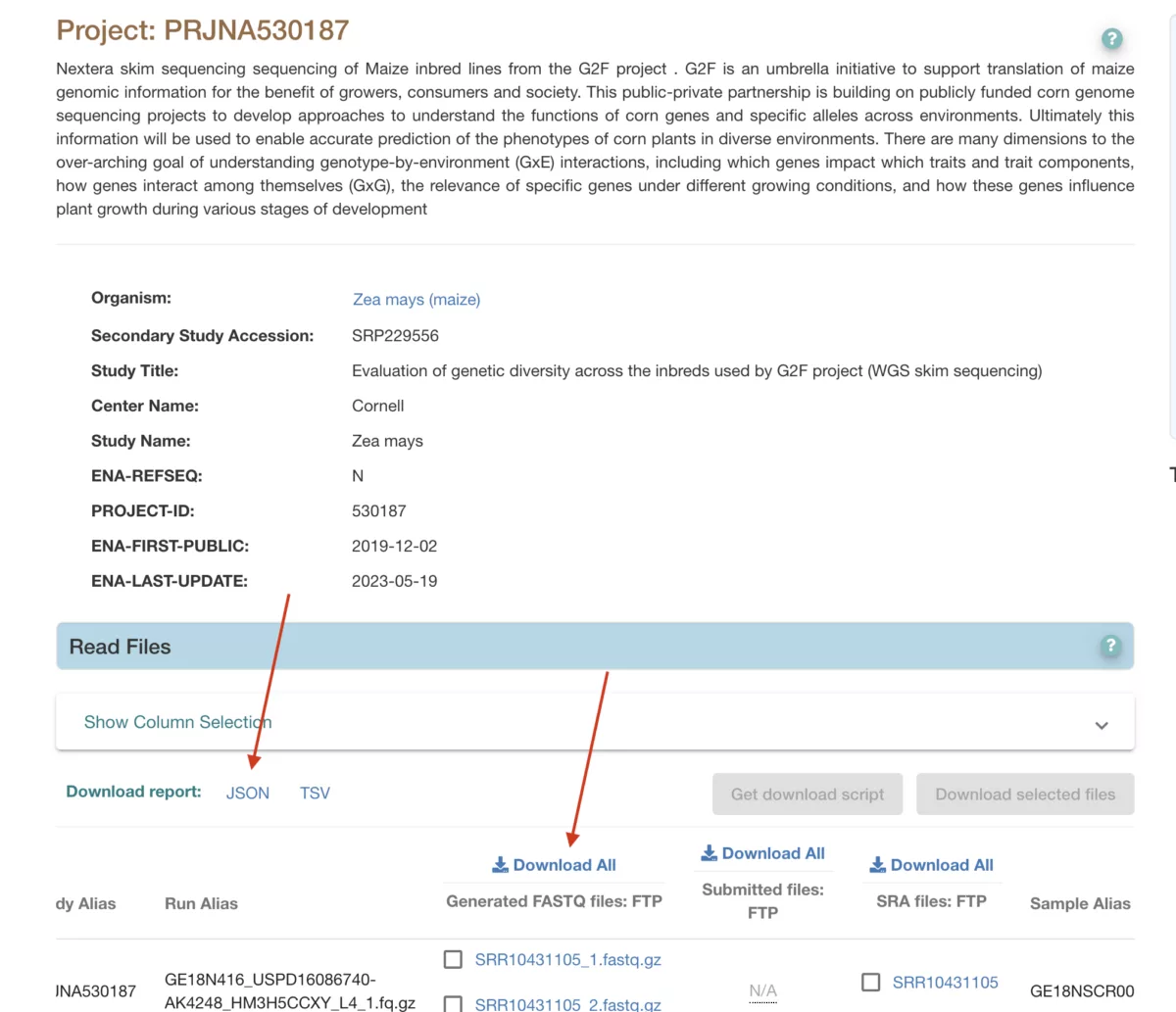
Here we will be using GenomesToFields (G2F) maize phenotype data here, and corresponding low-pass whole genome sequence data (approx 1-3x genomic coverage per line) from NCBI Project PRJNA530187.
The following code will be executed on a Gencove Explorer instance; through the use of the Gencove Explorer SDK, we will submit a high compute job to the analysis cluster that accompanies each Explorer deployment for data retrieval and processing, before downloading the results to our local instance in order to perform further analysis.
At a high level, we will perform the following steps:
- Retrieve and clean the G2F maize phenotype data (grain yields).
- Download low-pass whole genome sequence data from NCBI SRA for the same maize lines.
- Upload the sequence data to the Gencove SaaS for imputation.
- Prepare and clean the resulting imputed genotypes for genomic prediction.
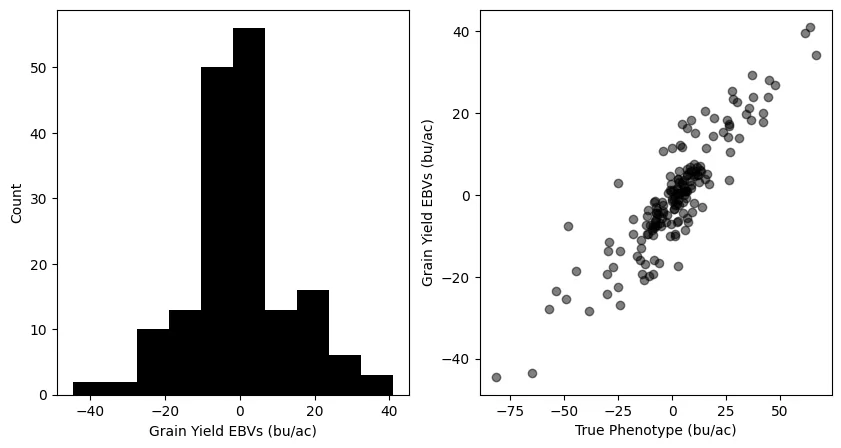
- Fit a Genomic Best Linear Unbiased Prediction (GBLUP) model using GEMMA on the Explorer analysis cluster
- Explore and visualize the results
- Upload the results to Explorer cloud storage.