The human leukocyte antigen (HLA) locus is a cluster of genes on chromosome 6 of the human genome that play a crucial role in self/non-self recognition in the immune system. Understanding genetic variation at the HLA locus is essential in human genetics as these genes are frequently associated with GWAS hits for common diseases and are crucial for organ transplantation1. However, accurately genotyping the HLA locus has proven challenging because it is complex and highly polymorphic.
A common approach for genotyping the HLA locus is to use imputation. Traditionally, an individual with an unknown HLA genotype is first genotyped using a backbone of biallelic markers in the HLA region, for example, from a genotyping array. These marker genotypes are then compared to a reference panel with known HLA genotypes, and the unknown individual’s HLA genotype is imputed by various means (e.g., machine learning2 or hidden Markov model3).
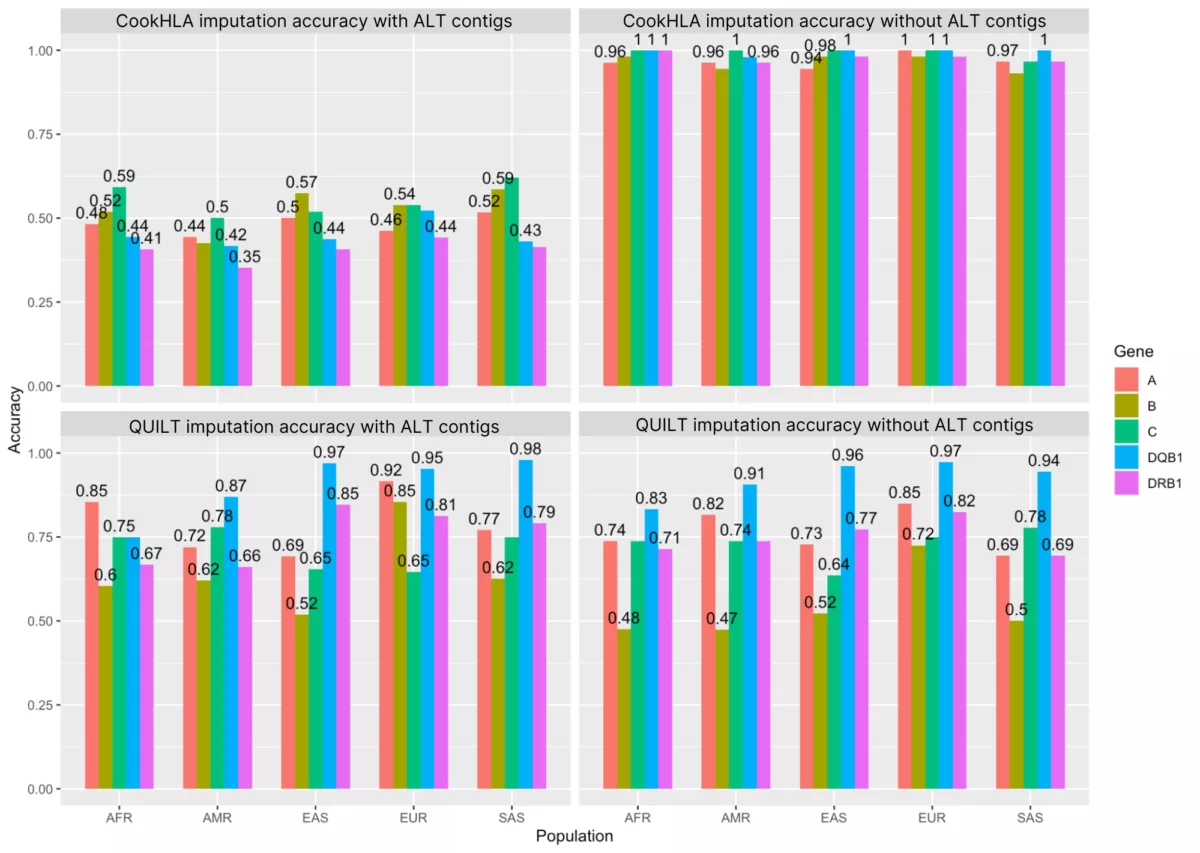
To adapt this method for use with low-pass sequence data, one would first need to impute the raw low-pass read data to a backbone set of markers and then use those markers to impute the HLA genotype. This “double imputation” approach has been shown to achieve high accuracy4. In this study, we use CookHLA3 for double imputation. CookHLA is a modern HLA genotyping algorithm that uses a hidden Markov model to achieve high performance.
Recently, a new approach called QUILT5 has been developed. Using a hidden Markov model, QUILT imputes the HLA region directly from raw low-pass reads, simplifying the double imputation method above.
Here, we compare these two HLA imputation approaches to learn which path leads to greater accuracy. Moreover, we evaluate two versions of the GRCh38 human reference genome. First, the primary release includes >500 HLA “ALT contigs” that represent diverse HLA alleles. Second, there is a special release of GRCh38, which lacks all ALT contigs, including those from the HLA region. Since these two assemblies differ substantially in how they represent the HLA region, we also looked at how the choice of GRCh38 reference genome impacts HLA imputation performance.