Overview
In the following blog, we will demonstrate how to perform analyses of population scale genomic data using Gencove Explorer. Broadly, we will explore fine-scale population structure in the form of rare variant sharing between samples deriving from the high-depth whole genome sequencing (WGS) dataset of the combined 1000 Genomes Project (1KG) and Human Genome Diversity Panel (HGDP) released as part of gnomAD v3.2.1.
The following code will be executed on a Gencove Explorer instance; through the use of the Gencove Explorer SDK, we will submit a high compute job to the analysis cluster that accompanies each Explorer deployment for data retrieval and processing, before downloading the results to our local instance in order to perform further analysis.
At a high level, we will perform the following steps:
- Retrieve large multi-sample whole genome sequence datasets pre-stratified by chromosome, and concatenate these into a single multisample VCF representing genetic variation across all samples genome-wide.
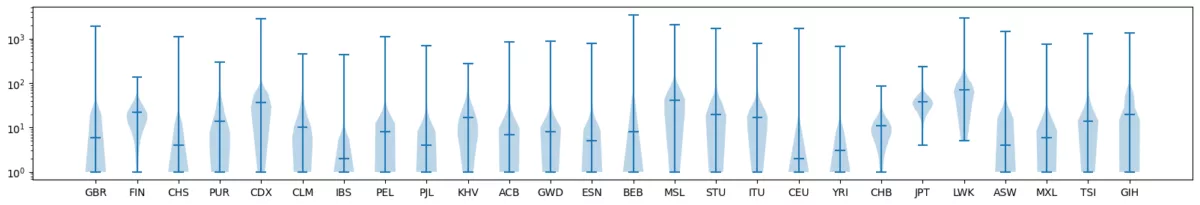
- Extract observations of doubleton sharing across all samples (i.e. instances of genetic variation observed only twice across all samples within the dataset).
- Retrieve the output of the above to our local instance.
- Generate a network based representation of doubleton sharing across the entire dataset.
- Visualize the distributions of doubleton sharing within population groups.
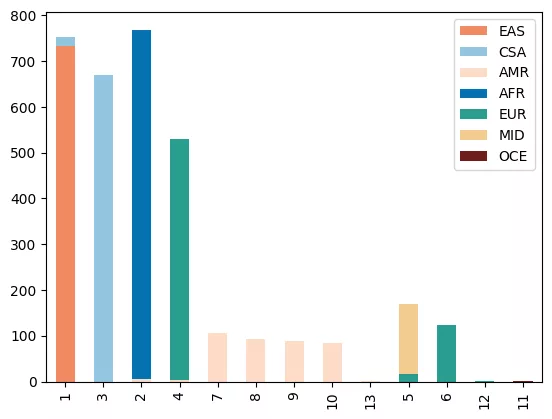
- Perform unsupervised machine learning over the network of doubleton sharing in order to explore patterns of recent ancestry within the data.
Introduction
Rare variants comprise the most frequently observed type of genetic variation in human genomes, and have the potential to be informative in genomic studies. Doubletons are a particular class of rare variation defined by being observed only twice across a population of samples. Previous work has demonstrated that the pairwise sharing of such variants between individuals can reflect patterns of recent shared demography, and in turn fine-scale genetic population structure (Mathieson and McVean, 2014). The demographic information revealed by the analysis of rare variation has broad applications across a variety of sub-fields of human genetics, including adjusting for population structure in genome-wide discovery efforts (Persyn et al. 2018), accounting for stratification in the calculation of polygenic risk scores Zaidi and Mathieson, 2020, understanding patterns of segregation of clinically relevant variation, and elucidating historical relationships between groups via genomic analysis (Gravel et al 2011).
The ascertainment of genetic variation on genome-wide arrays typically prohibits the analysis of rare variant sharing in an unbiased manner; however with the recent increase in public availability of whole genomes captured via deep sequencing, there are emerging opportunities to explore and further characterize rare variation at the population scale.
Leveraging the Gencove Explorer platform, we will demonstrate how one might characterize patterns of rare genetic variation in population scale whole genome sequence data, and apply unsupervised machine learning in order to extract insights from patterns inherent within the data. We do this by utilizing the 1000 Genomes Project and Human Genome Diversity Panel (Koenig et al. 2023) a globally diverse population-scale dataset comprised of N=4094 whole genomes covering n=77 population groupings, and encompassing over 153 million genetic variants,released as part of GNOMAD v3.2.1.
Environment Setup
First, we will start by importing a few key objects from the Gencove Explorer SDK. These will allow us to easily interface between the Explorer deployment's analysis cluster, cloud storage, and our local instance, allowing for easy file access and job submission (for more detailed information on Gencove Explorer and its associated functionalities, please refer to the Explorer documentation here):