At Gencove, we’re continuously expanding the selection of species which can be used with imputation pipelines based off of low-pass sequencing. Unlike, for instance, cattle in the form of the 1000 Bull Genomes Project [1], there does not currently exist a large-scale public resequencing effort to characterize the genetic diversity of extant breeds used for agricultural purposes, and the existing literature on the performance on genotype imputation in pigs is primarily limited to genotyping array imputation [2,3,4].
To address this current shortcoming, we have recently developed and released a pipeline for low-pass sequencing in pigs on the latest reference genome (Sscrofa11.1/susScr11) and a diverse haplotype reference panel encompassing the range of genetic diversity in the most common breeds found in the swine industry.
This panel enables both low-cost genotyping for genomic selection as well as an unprecedented level of resolution for a high throughput assay for research and fine-mapping in swine.
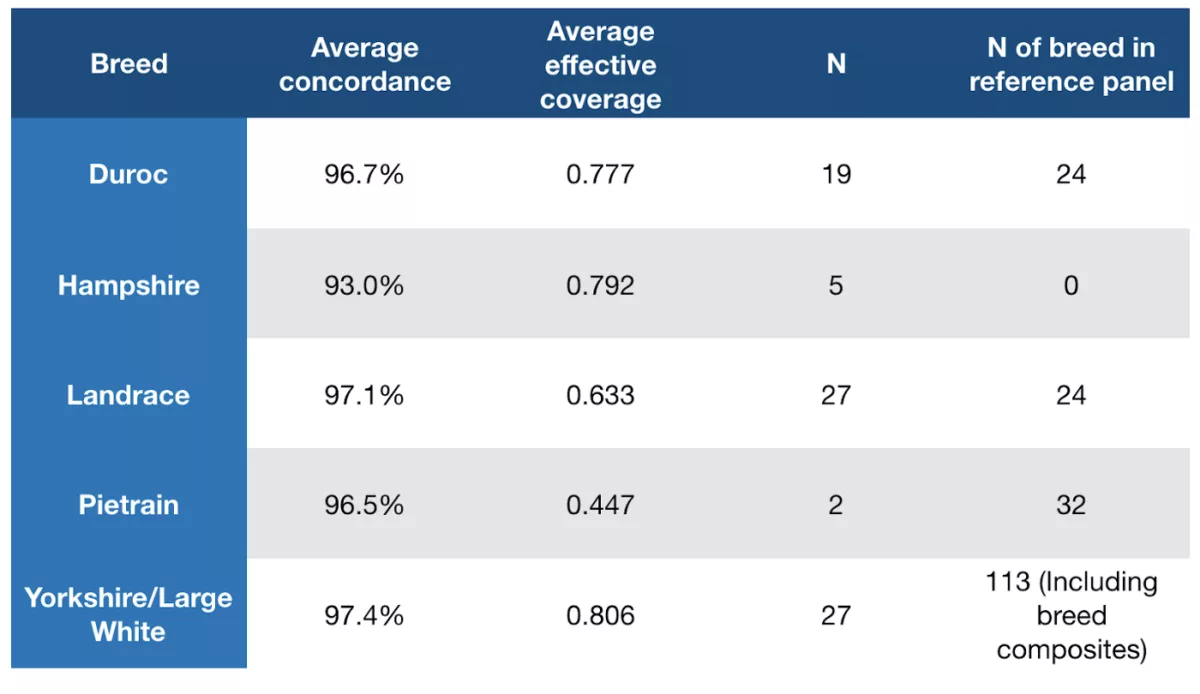
In order to test the performance of our imputation pipeline, we performed a pilot in partnership with the USDA-ARS Meat Animal Research Center (USDA-ARS MARC) wherein we sequenced and imputed 83 boar samples provided by the USDA-ARS and compared the imputed genotypes to those deriving from an orthogonal assay, the GGP Porcine 50K array. Thanks to their historical public sequencing efforts, the MARC populations are relatively well represented in our panel, but as we will see below, even the subset of samples deriving from breeds not well-represented in our panel imputed reasonably well due to the large amount of shared genetics between pig breeds.
Haplotype reference panel construction
To construct our haplotype reference panel, we first collated publicly available swine sequence data on the Sequence Read Archive (SRA) from 414 individuals. We then created a haplotype reference panel using our in-house reference panel building pipelines that have been applied and validated on a number of other agricultural (and human) species. The resulting reference panel comprises 53M SNPs and short indels.
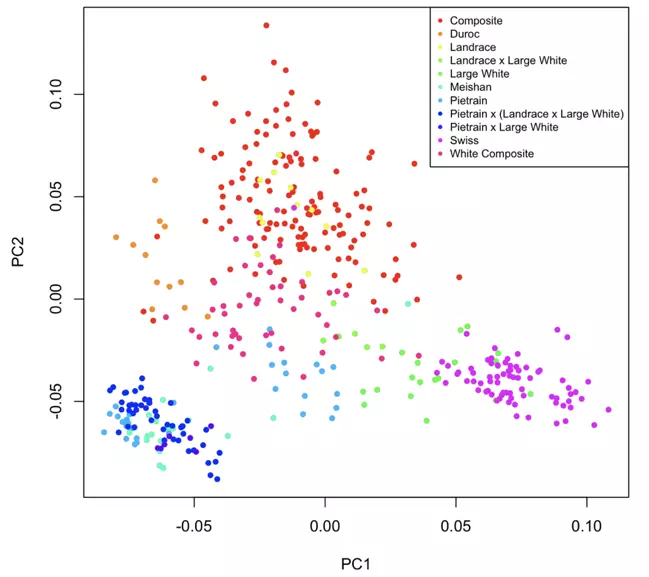
To examine the population structure of the samples comprising our reference panel, we performed PCA on a subset of the markers in our panel. In the following figure, the axes are the first two principal components of the marker subset, and each point on the plot represents an individual in our reference panel. Each point is colored by the sample breed, and animals of the same breed can be seen to cluster together, illustrating the distinctness of population groups. These lines span the full genetic diversity of publicly available data, and therefore ensure that any incoming sample sharing genetics with these individuals will be accurately characterized by imputation to this reference panel.