Over the past decade, human genomic research has benefited tremendously from the availability of open-access human genome diversity panels like the 1000 Genomes Project (1KG) and the Human Genome Diversity Project (HGDP). Both projects released high-quality genome sequences from a diverse set of individuals. Though this data is readily available, it has proven to be a challenge to combine and harmonize variant calls between these panels to make a single high-quality dataset.
In a blog post earlier this year, we used low-pass imputation performance to evaluate a combined HGDP + 1KG panel (HGDP1KG) produced by a team led by Dr. Alicia Martin at the Broad Institute. This combined panel was compared to the expanded 1KG panel released by the New York Genome Center (NYGC1KG). Though the combined HGDP1KG panel was larger, we found that the NYGC1KG panel had higher imputation accuracy, especially at indel sites.
Recently however, the team at the Broad released an update to the HGDP1KG panel as part of the gnomAD_v3.1.2 release that included several tweaks and changes meant to increase the accuracy of the combined panel. In this blog post we once again compare imputation performance of this updated panel to NYGC1KG panel.
Panel Comparisons
The new version of the HGDP1KG comprises 4091 individuals with ~76.4M variants (~67.2M SNPs and ~9.2M indels) among the 22 autosomes, while the NYGC1KG dataset comprises 3202 individuals and contains ~73.6M variants (~64.1M SNPs and ~9.5M indels) among the autosomes and X chromosome. This latest release of the HGDP1KG is considerably smaller than past releases. For example, we found that the past release of the HGDP1KG contained roughly twice as many SNPs as the NYGC1KG. Due to filtering performed on the gnomAD_v3.1.2 release, it now has a comparable number of variants to the NYGC1KG panel.
Results
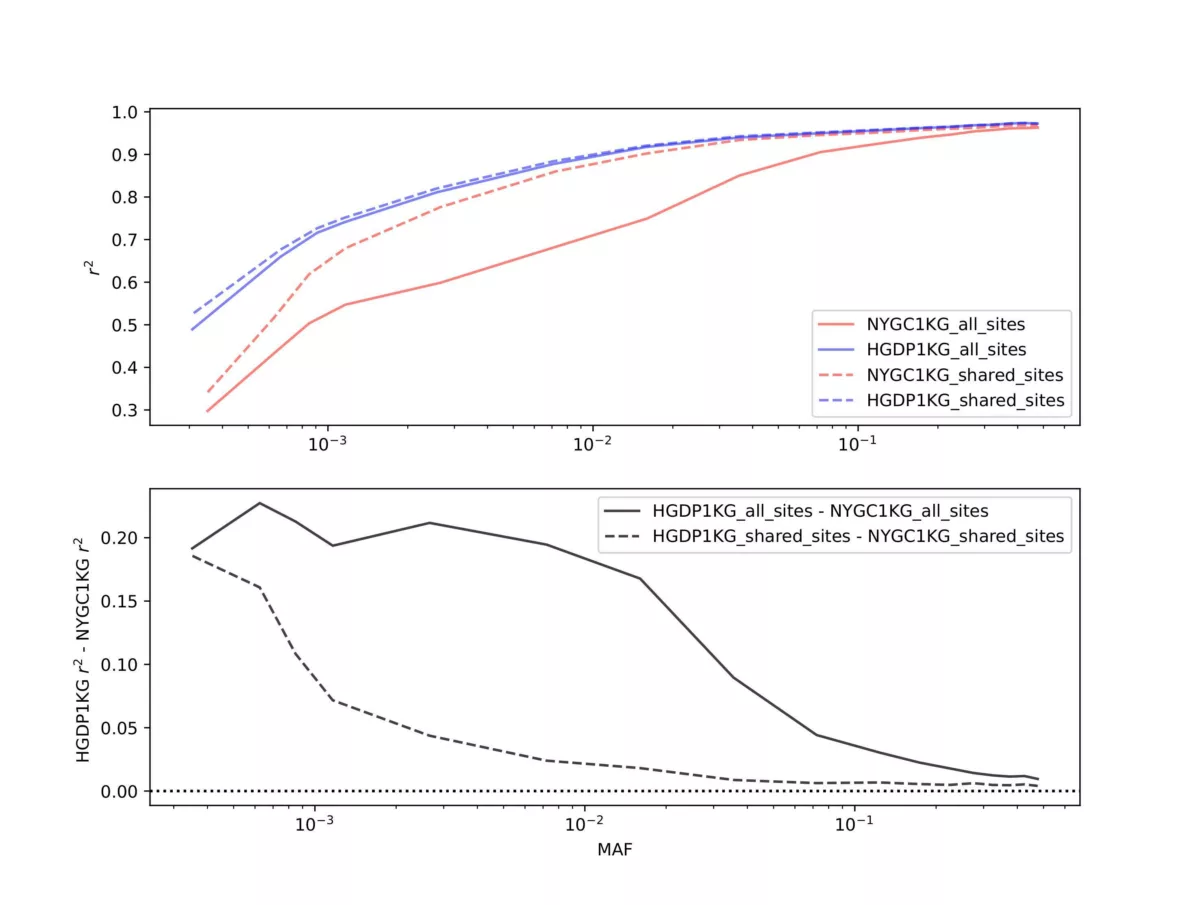
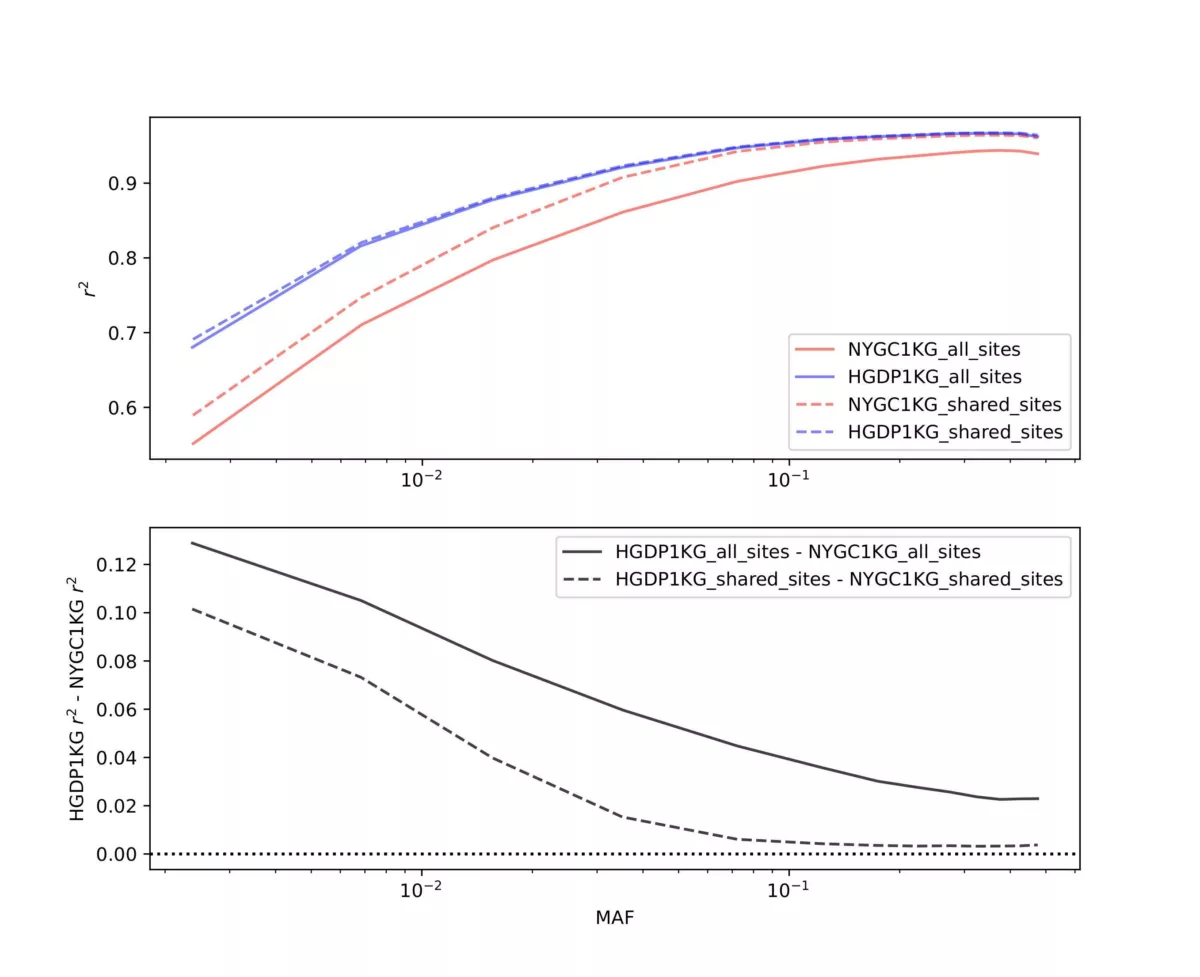
Two panel comparisons were performed. First we selected 60 individuals that are found in both the NYCG1KG and HGDP1KG panels and imputed them using GLIMPSE2 with either panel in a “leave-one-out” manner, meaning the target individual was removed from the reference panel, and downsampled to 1x coverage for imputation. For efficiency we only performed this analysis on chromosome 20. This same design (leave-one-out on the same 60 individuals for chromosome 20) was used in our recent preprint. Figure 1 contrasts panel performance. The metric we evaluated was the imputation r2, which is the squared Pearson correlation coefficient between the imputed genotype and the truth genotypes provided by the high coverage genotype calls. The aggregate imputation r2 among all 60 individuals is stratified by minor allele frequency (MAF) of the site in the respective reference panel. In the upper panel of Figure 1 the blue lines represent the HGDP1KG panel while the red lines represent the NYGC1KG panel. Solid lines represent the imputation r2 performance when considering all sites in the panel, while dashed lines represent imputation r2 performance at sites shared between both reference panels (~1.4M sites) on chromosome 20. The lower panel of the figure represents the imputation r2 difference between the HGDP1KG panel and the NYCG1KG panel results, again stratified by MAF.