Whole exome sequencing (WES) is a popular and cost-effective method for capturing high quality genotype calls at the protein coding portion of the genome, and has been key in facilitating the discovery of functionally relevant variants across a range of research and clinical settings.1-7 Despite this, WES is designed to only capture exonic regions, meaning that the yield of on-target calls obtained through this technology is typically restricted to only around 1% of the genome in total. This has the potential to hamper discovery efforts that solely rely on WES, as it means that the intronic and intergenic fraction of the genome, which comprise the majority, remain un-assayed. Notably, over 90% of signals discovered through Genome Wide Association Studies (GWAS) fall within non-coding regions of the genome,8 and many analytical methods in the field of genomics rely on dense sampling of genotypes across samples genome-wide for statistical inference. Consequently, researchers who choose to employ WES for large-scale discovery efforts often have to supplement exome sequencing with array-based genotyping technologies in order to capture a more complete picture of their study population.
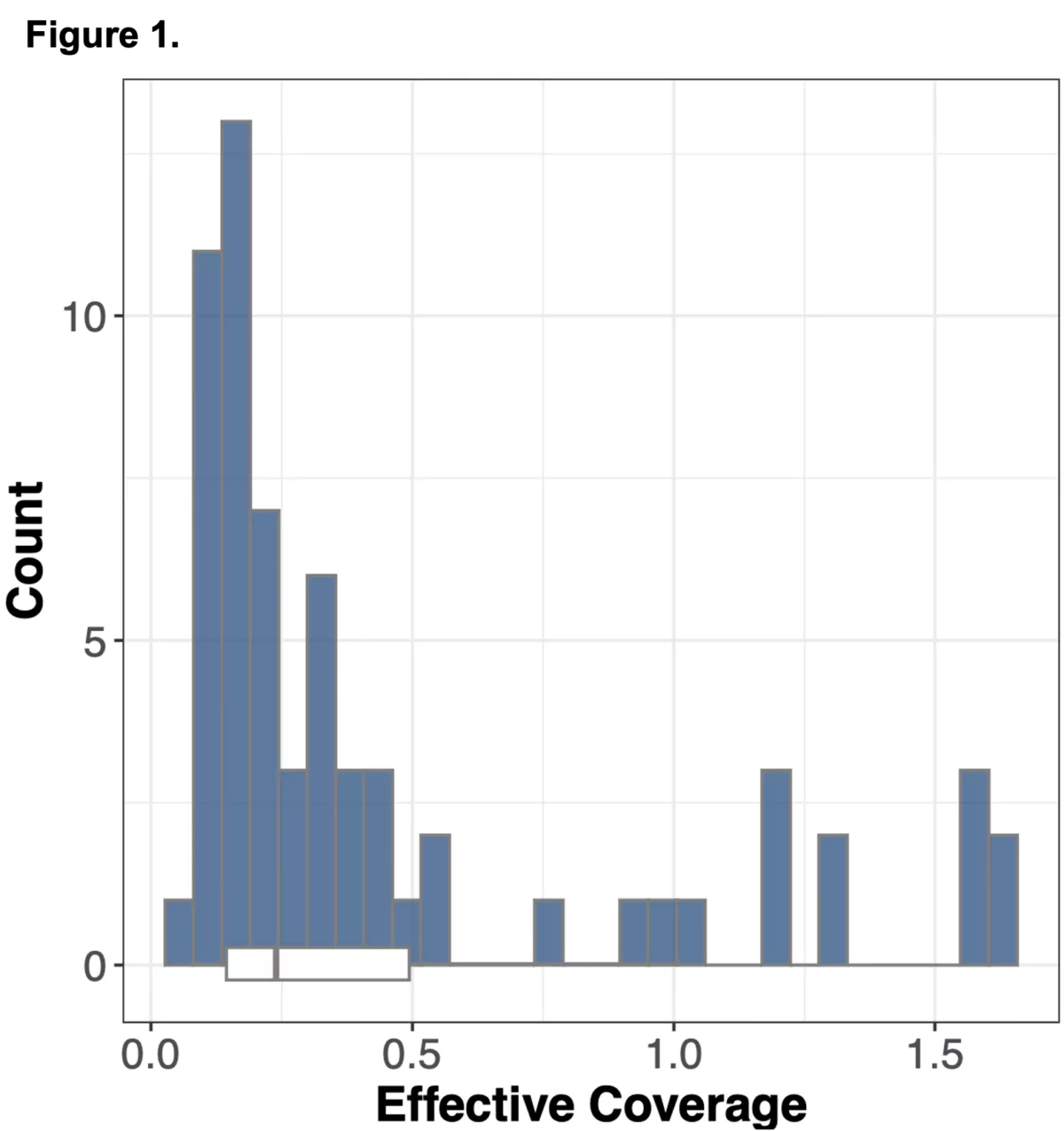
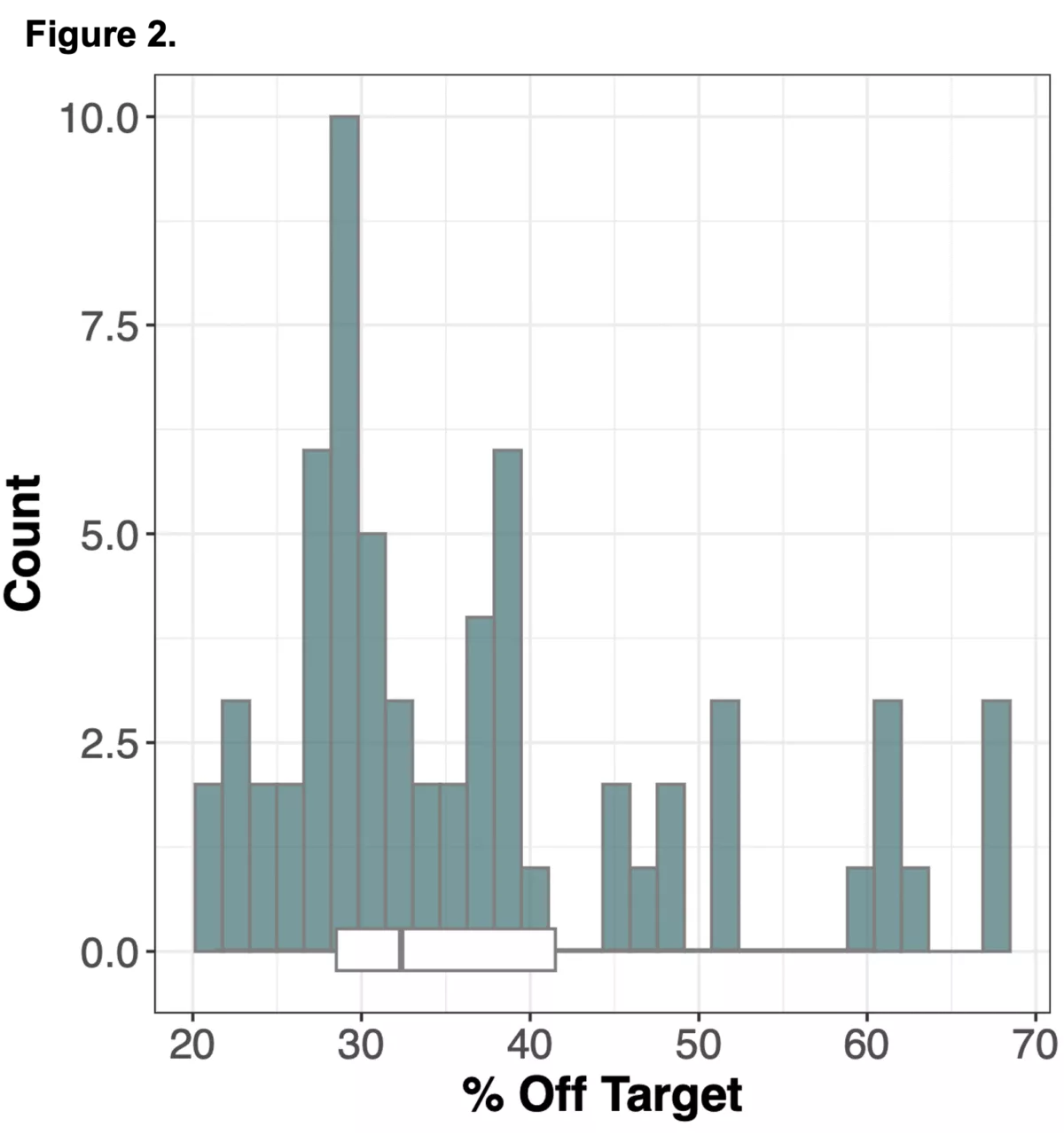
We wanted to explore an alternative approach to obtaining genomic information in non-coding regions of the genome using WES alone, by leveraging information from off-target reads9. Off-target reads refer to sequencing data that is captured from regions outside of the intended exonic targets. Although WES is specifically designed to enrich and sequence the exonic regions of the genome, the hybrid capture method used is not perfectly selective, leading to the incidental capture of sequences from non-exonic regions. The proportion of off-target reads in a standard WES study ranges from 20-40%10 of the total read yield, and while typically not incorporated into downstream analysis in standard WES workflows, has the potential to be repurposed for assaying genotypes across the whole genome.
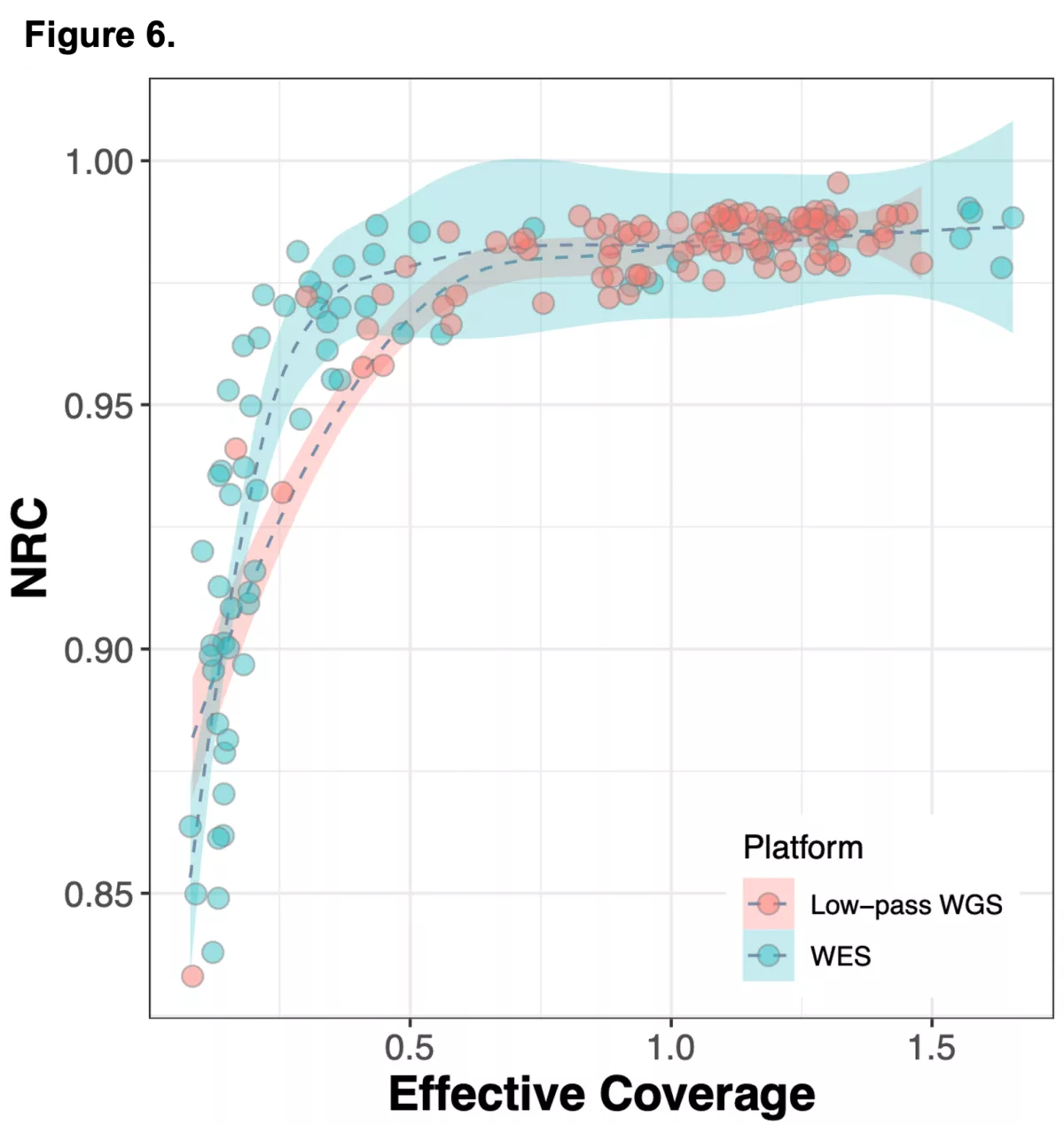
We sought to combine information from off-target WES reads with state-of-the-art imputation methods, typically used for low-pass whole genome sequencing (lp-WGS), to see if we could obtain high quality genotypes at non-coding regions, providing both high coverage exome calls and genome-wide genotypes in one assay.
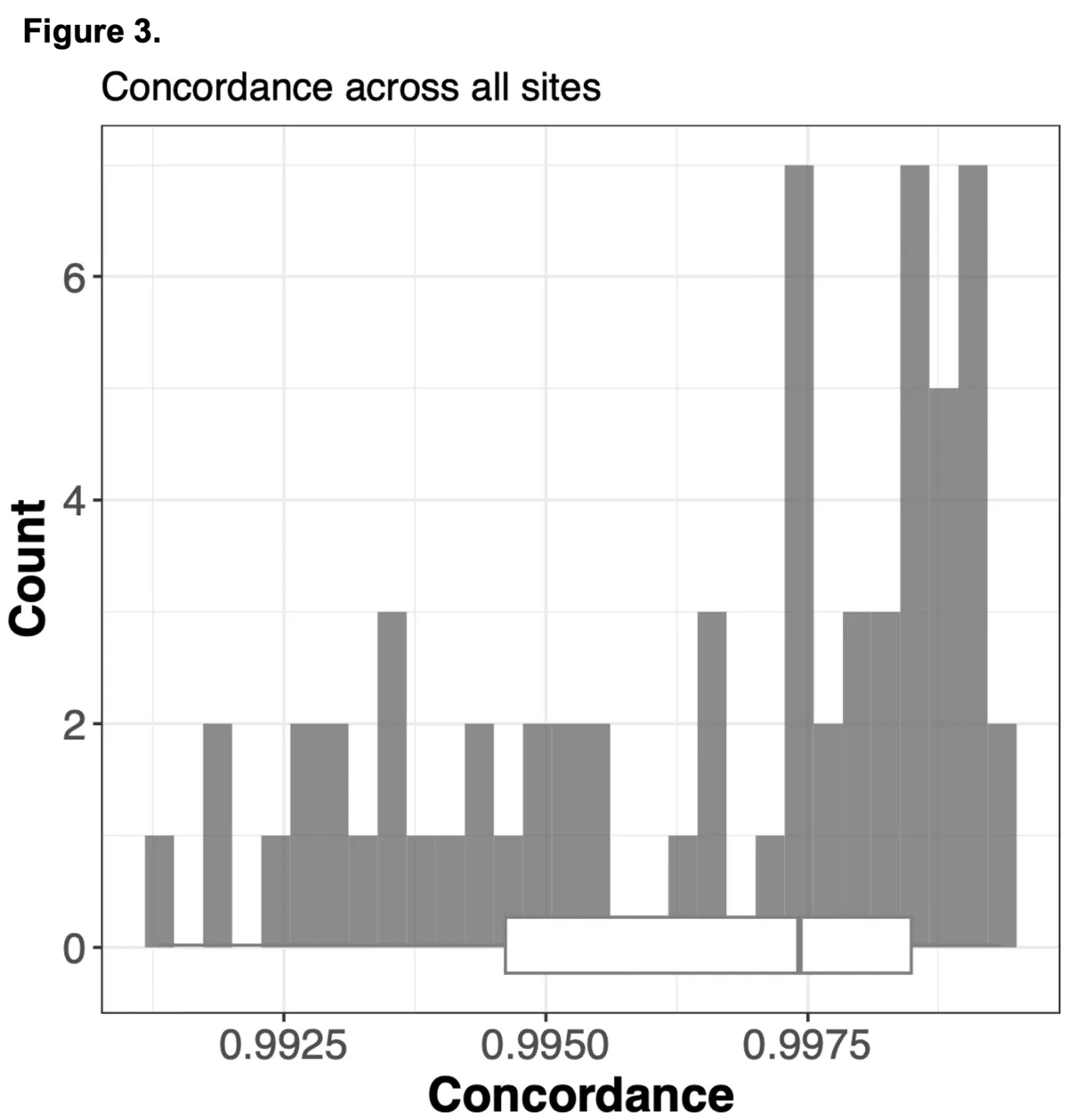
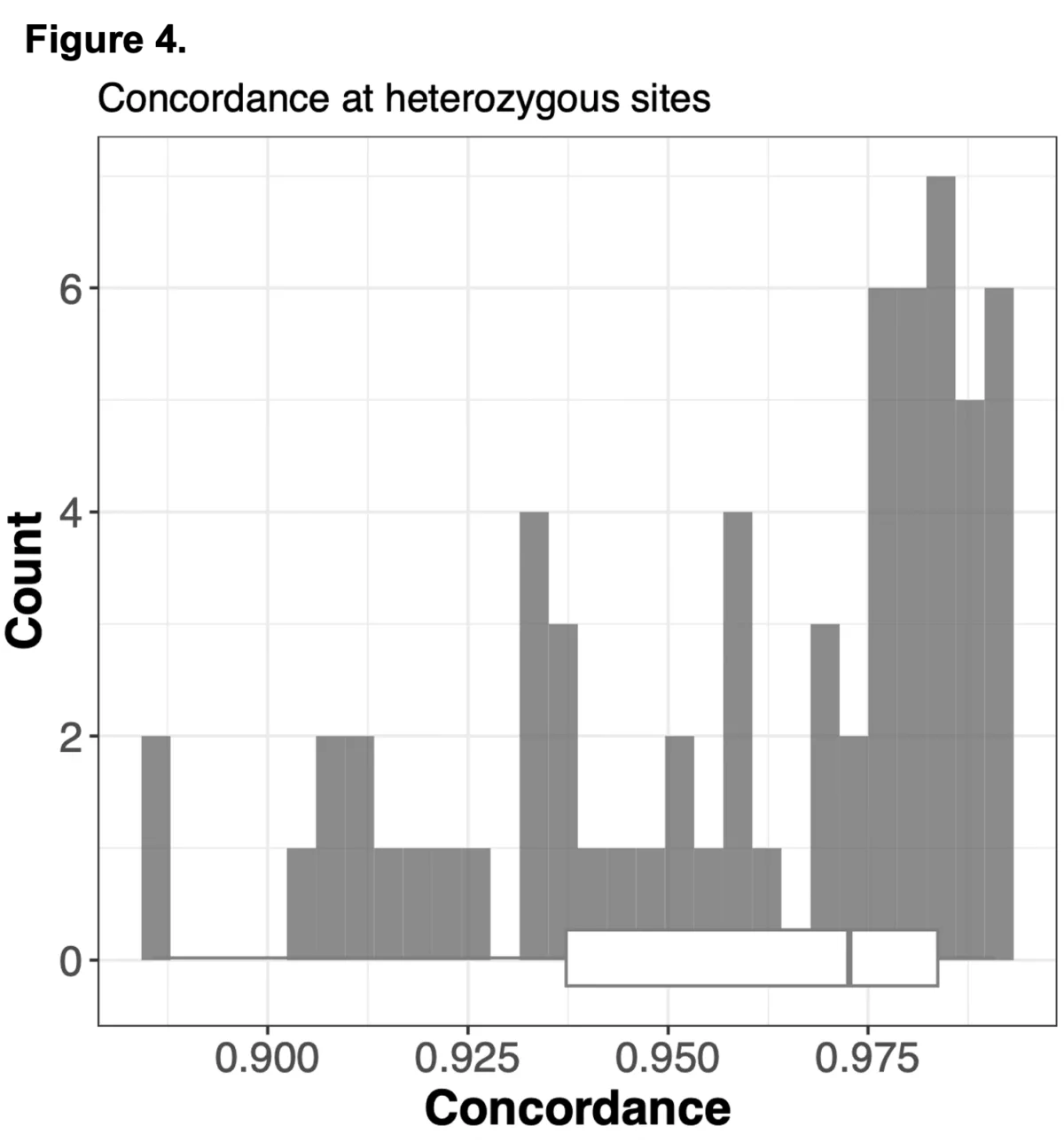
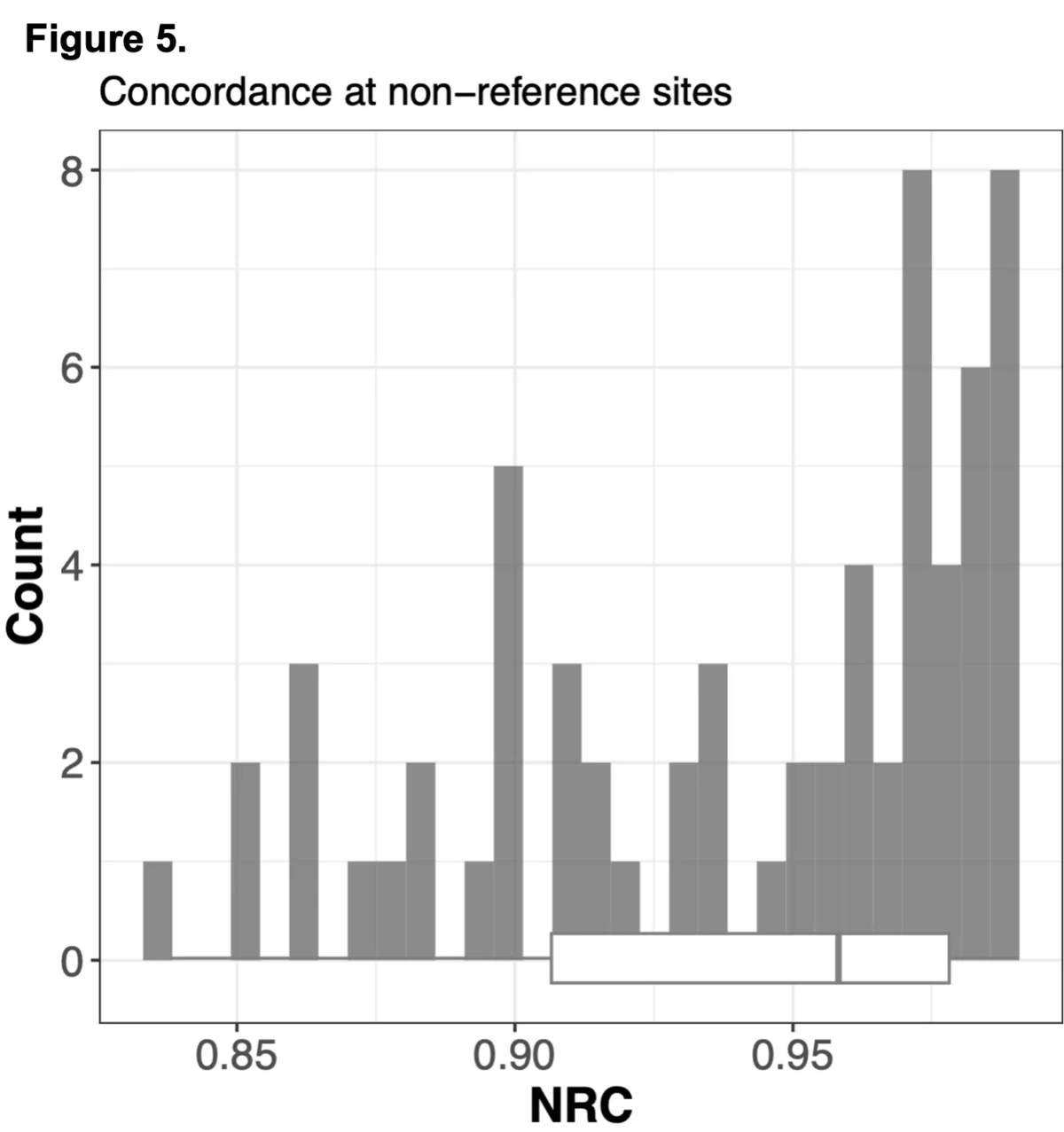
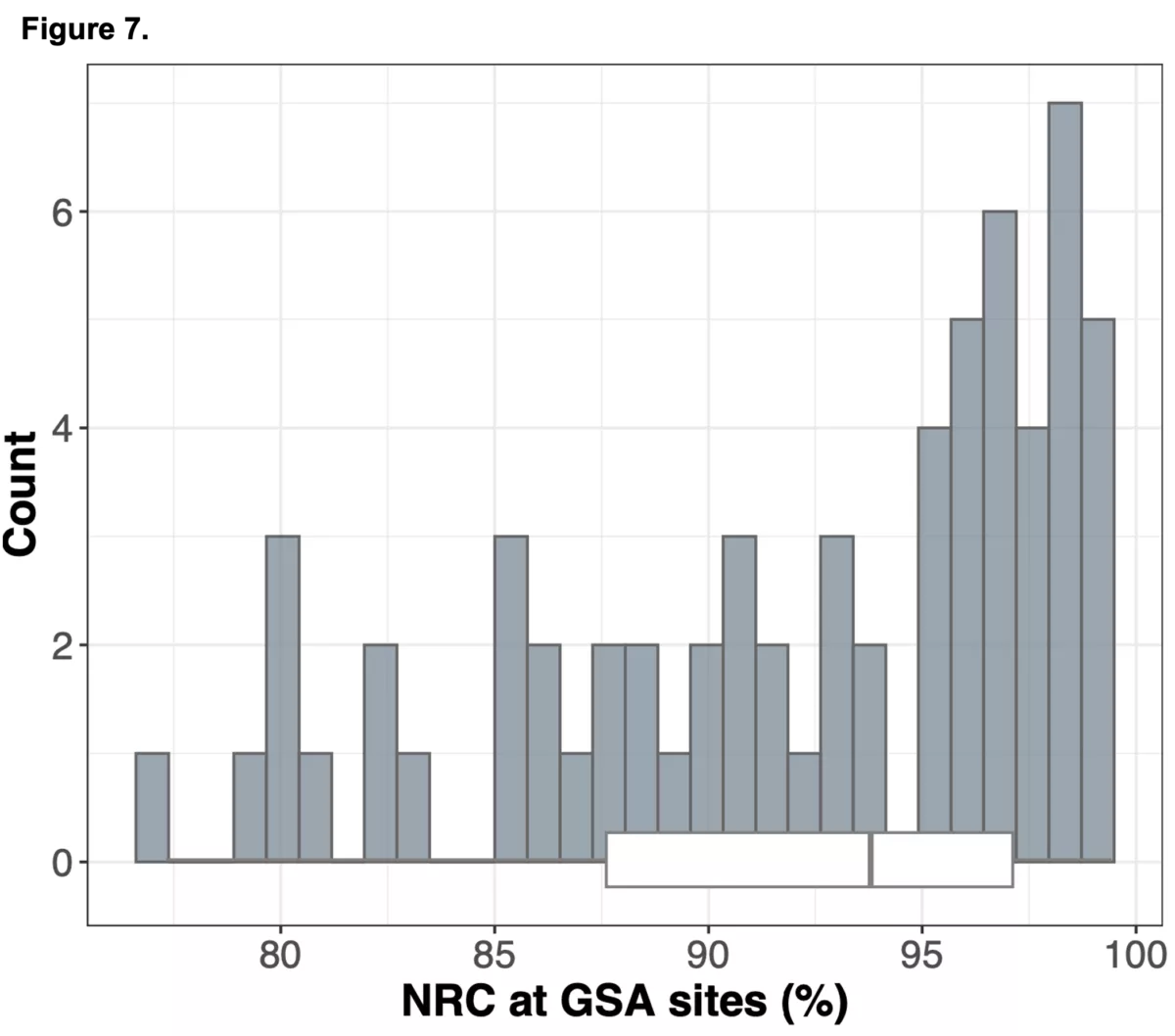
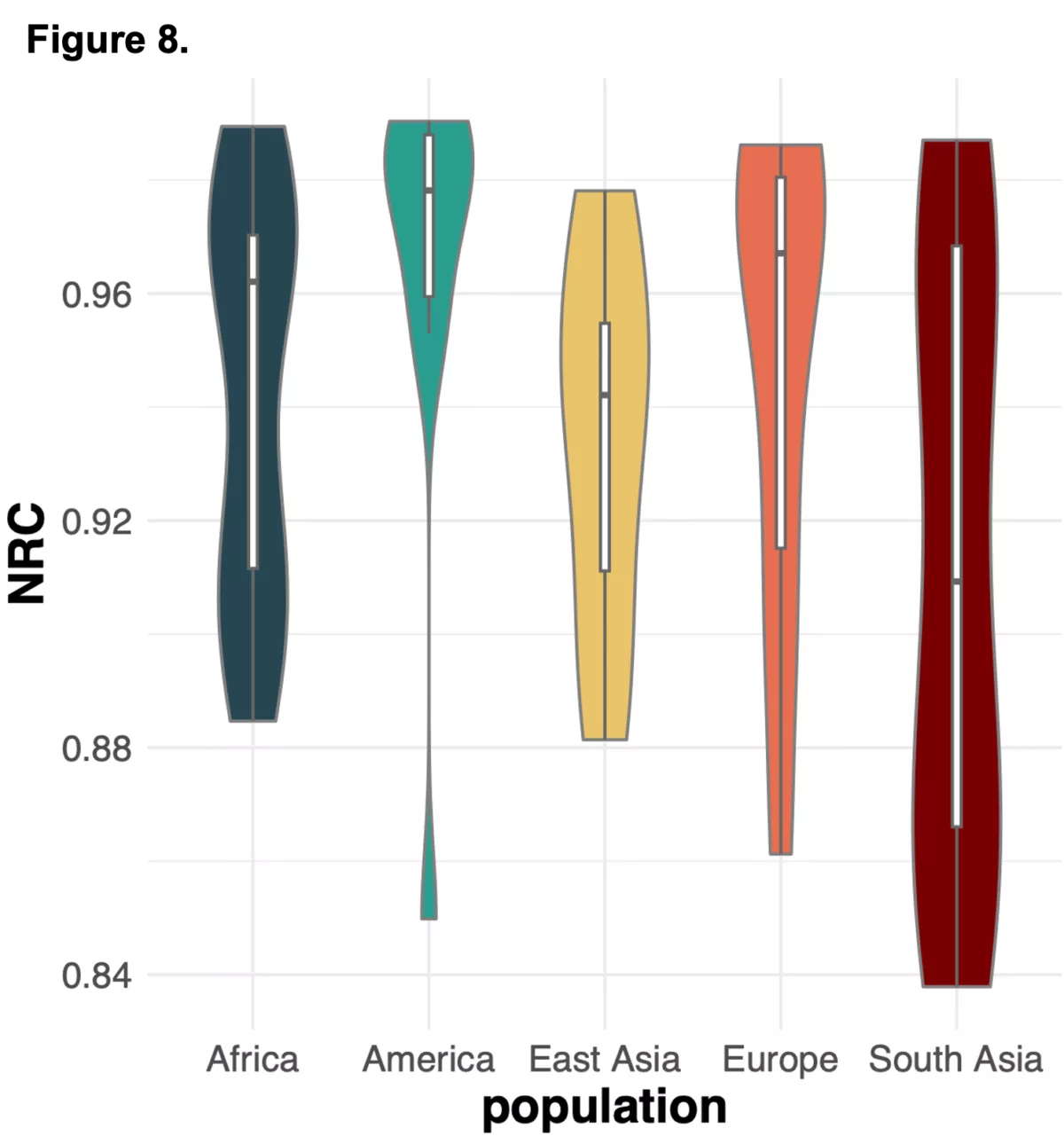
To do this we analyzed N=64 samples from the 1000 Genomes Project (1KG) that are present in the Gencove imputation reference panel and for which high coverage WES data is available. We analyzed the WES samples using the Gencove imputation platform in a leave-one-out manner (that is, ensuring the exclusion of the test sample from the reference panel at the time of imputation). We were then able to benchmark the quality of the genotype calls obtained by comparing imputed calls to the “ground truth” high coverage whole-genome sequencing data that is publicly available for that sample.