The vision of Gencove is that the universal application of genomics will result in a healthier and more sustainable civilization.
As we work towards that vision, we regularly process large amounts of genomic information and it is commonly accessed from remote locations like object storage (e.g., AWS S3), for that purpose we’ve written a tool called htsutil for subsetting a VCF file outside of the HTSlib ecosystem.
We are huge fans of HTSlib/bcftools and use it extensively, but in our hands it was somewhat unreliable when fetching data from remote network locations in high-volume production settings.
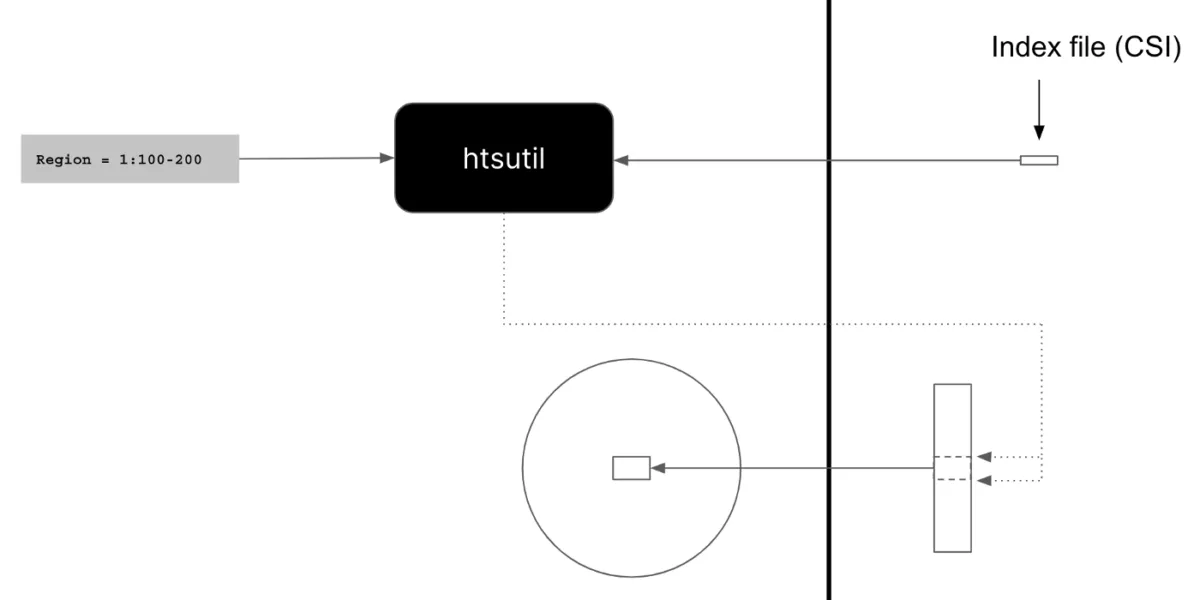
Htsutil takes a CSI index file and a region and returns the byte offsets that you need to read from a VCF file, which can be passed to downstream tools for efficient processing of VCF files.
This means we can now use downstream tools to:
- Robustly fetch information from our VCF files by streaming files from S3 using curl or AWS CLI.
- Efficiently fetch information from our VCF file by using the byte offsets that htsutil provides.
Also, we’ve released htsutil as an open source project that is available on Gitlab.
How does htsutil work?
Imagine you have a VCF file stored in a remote location and you need to use it inside an EC2 instance to run some analysis.