Over the past decade, the cost of genome sequencing has significantly decreased, leading to a rise in popularity of alternative sequencing methods such as low-pass whole genome sequencing instead of genotyping arrays. To meet this demand, at Gencove, we offer a cost-effective solution for generating genome-wide information by providing low-pass whole genome sequencing plus imputation as a high-throughput alternative to genotyping arrays. Our previous research has already established that low-pass whole genome sequencing outperforms traditional genotyping arrays in terms of imputation in humans (Li et al., 2021). In this blog post, we describe the results of our recent experiment where we demonstrate that imputation using low-pass whole genome sequencing is also superior to genotyping arrays in cattle.
Motivation:
The commercial use of genotyping arrays followed by imputation to a haplotype reference panel is a widely used method to generate genome-wide information in cattle for research purposes. However, despite their popularity, genotyping arrays have some inherent limitations that restrict their long-term usage. One such limitation is the pre-defined set of markers selected for inclusion in the array, which can lead to ascertainment bias. In contrast, sequencing technologies agnostically probe the entire genome and provide the potential to identify novel variation. For instance, the BovineSNP50 Array, a high-density genotyping microarray routinely used in cattle genetics, contains just over 51 thousand SNPs distributed uniformly across the genome of major cattle breed types. While these SNPs are highly informative, they provide only a snapshot of the genome that serves as the backbone for the imputation. In contrast, low-pass whole genome sequencing at a target coverage of as low as 0.5x provides a number of reads that are orders of magnitude higher than the number of sites assayed on traditional arrays. This gives far more observations of informative markers for the imputation algorithm which results in more accuracy across the genome. The typical low-pass sequence run at a coverage of ~1x results in measurements at millions of known polymorphic sites in the cattle genome.
Experimental Design:
In this experiment, our primary aim was to identify the inflection point at which low-pass whole genome sequencing surpasses genotyping arrays in terms of imputation performance in cattle. To evaluate this, we randomly selected 50 Holstein and 50 Angus and obtained high-coverage sequence data for them. We then generated low-pass data for these subjects by downsampling the high-coverage data to a range of coverage levels and masked the high-coverage data to the 50k sites on Bovine50SNP Array to simulate high confidence array data for imputation.
In total, we generated seven sets of input data for imputation as follows:
Simulated Bovine50SNP Array (51,110 sites)
0.1x
0.5x
1x
2x
3x
4x
Next, each set of input data was imputed to our largest cattle haplotype reference panel in a leave-one-out manner using IMPUTE5 for the simulated array data and GLIMPSE for low-pass sequence data. These two state-of-the-art imputation algorithms are expected to provide the most robust imputation results for array and low-pass data, respectively.
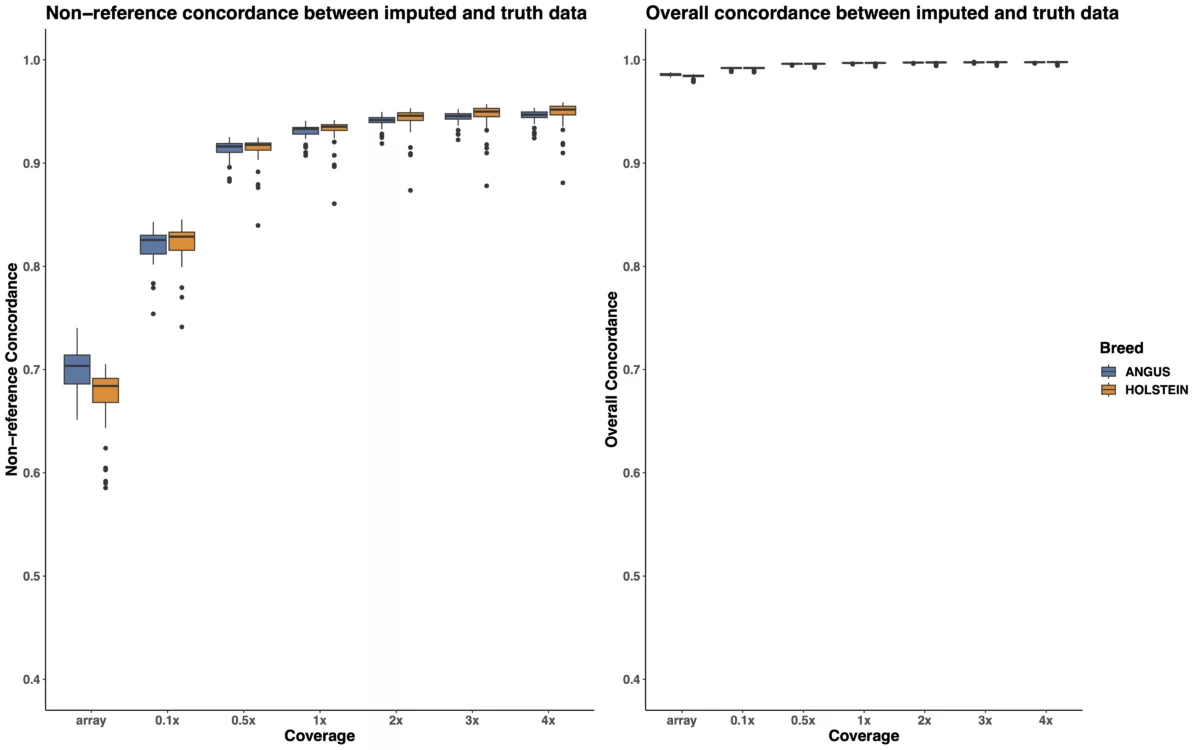
To examine the relative performance of imputed low-pass and array data, we compared the imputed genotypes for each run against the gold standard truth genotypes obtained from our high-coverage reference panel and calculated overall and non-reference concordance estimates for each run. In the context of this experiment, overall concordance refers to the accuracy with which all imputed variants match the gold standard truth genotypes in the reference panel, while non-reference concordance refers to the accuracy with which imputed non-reference variants match the gold standard genotypes for non-reference variants in the reference panel. Because of the high number of reference genotypes included in the overall concordance estimation, overall concordance is generally expected to have less sensitivity while non-reference concordance has better discrimination power for determining the relative quality of imputation for each run.
To provide some additional context, our current cattle reference panel used in this experiment includes 171,774,809 sites from 2,293 samples with high-coverage sequence data that spans a large range of genetic diversity in cattle.
Results:
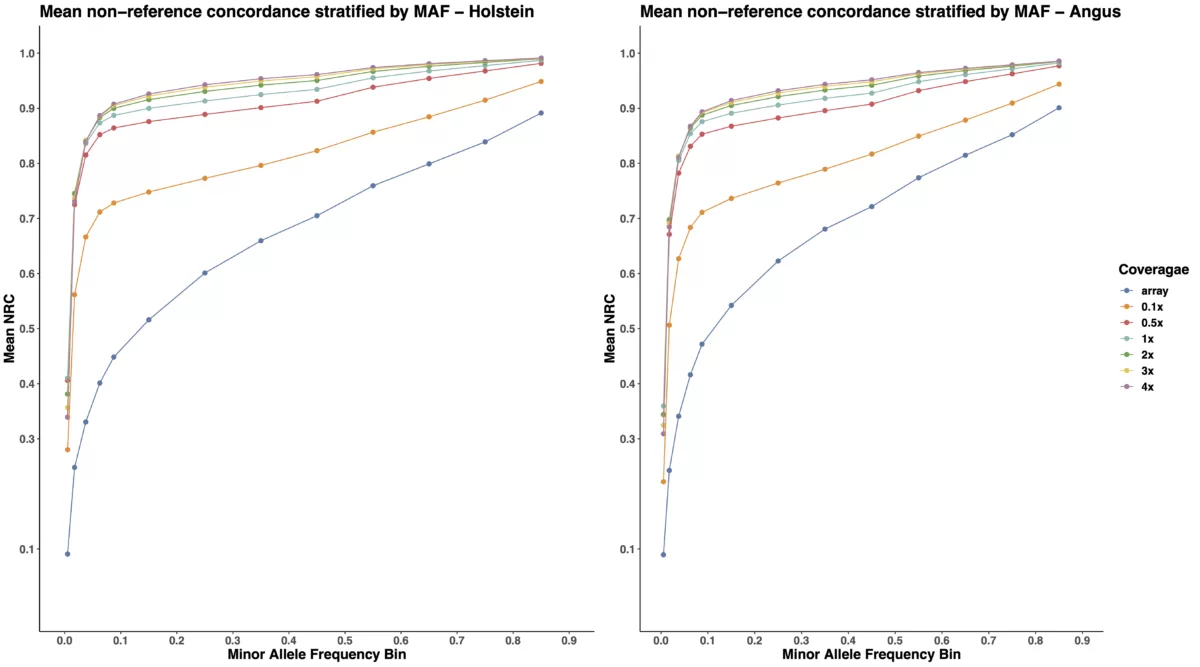
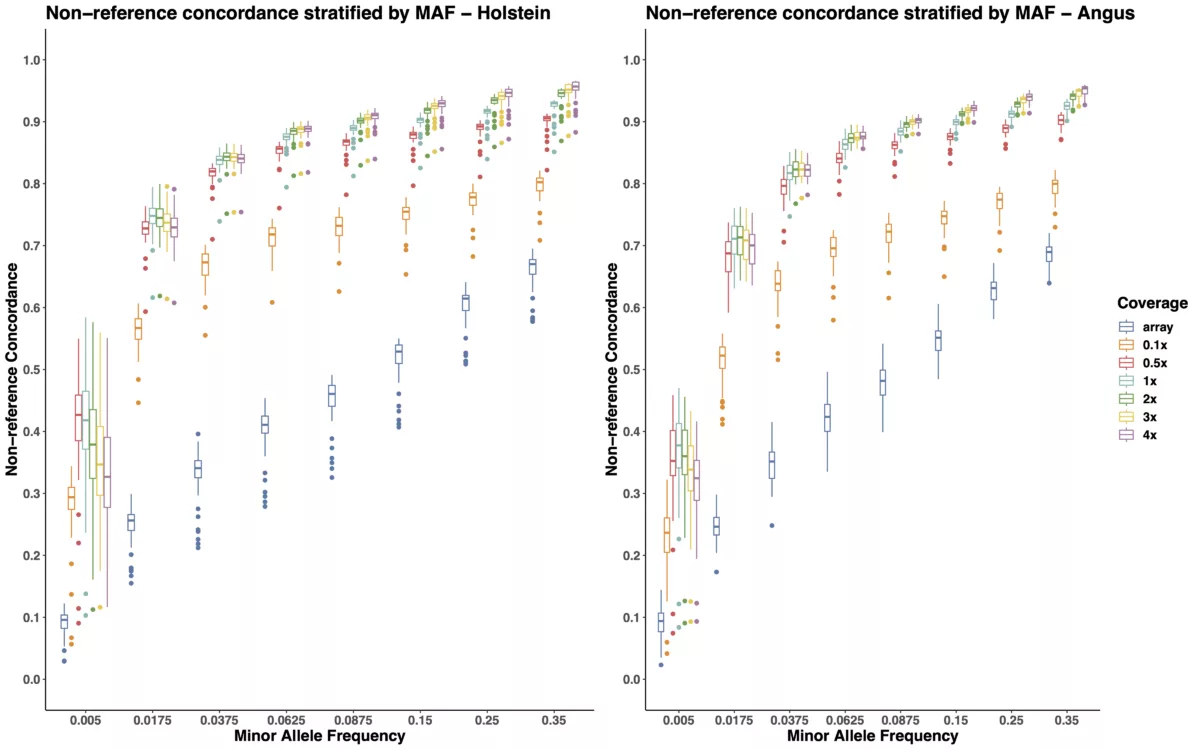
The boxplots in Figure 1 demonstrate that low-pass imputation at a coverage as low as 0.1x is superior to array-based imputation, in terms of both overall and non-reference concordance estimates. As shown in Table 1 and the left panel of Figure 1, the mean non-reference concordance for array-based imputation is 0.6859, whereas the mean non-reference concordance for the shallowest coverage of low-pass imputation (0.1x) is 0.8216. While increasing the coverage from 0.1x to 0.5x provides demonstrable improvement in imputation quality by increasing the mean non-reference concordance from 0.8216 to 0.9133, we note that increasing the coverage to higher than 0.5x results in quickly diminishing returns in terms of imputation accuracy, suggesting that 0.5x is likely to provide adequate coverage for most applications in cattle. Furthermore, the trend we observe here is consistent across two distinct cattle breeds, Angus, and Holstein.