What exactly is low-pass sequencing? It has gone by many names since it emerged as a viable alternative to microarrays in 2012, including low-pass or low pass sequencing, low coverage sequencing, and skim sequencing. In general, low-pass sequencing refers to whole genome sequencing (WGS) at a much lower coverage (typically defined as less than 1x coverage) than standard WGS and using statistical analysis, or imputation, to call SNPs based on known genetic variation within the population
Blog
Gencove Team - Aug 10, 2023
Low-pass sequencing and imputation for evaluating genetic variation
Why consider reducing the coverage?
Whether you’re breeding cattle or screening patients for pharmacogenomics, the traditional way to genotype individuals is to use a microarray. While microarrays have reduced the cost to perform genotyping at large scale, they can only deliver a known subset of genetic variants across any given genome, limiting the information you can gain to just those specific polymorphisms that have been built into the array.
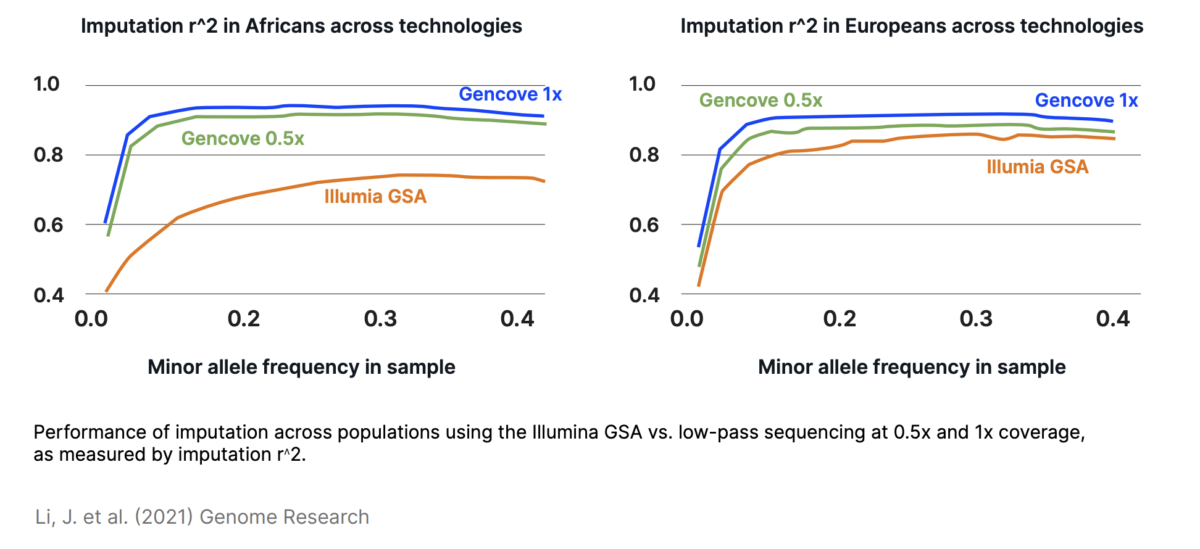
Wasik, et al. in 2021 compared microarrays and low-pass sequencing performance in the human genome. “As an intuition for why [low-pass] is useful, note that a human sample sequenced at 0.4x coverage is expected to have a single sequencing read covering each of around 28 million of the 84.7 million genetic variants identified in the 1000 Genomes Project, while a genotyping array obtains measurements… at two orders of magnitude fewer sites,” state the authors. Therefore, if the decision is between low-cost microarray and information-rich WGS, low-pass sequencing provides a practical way to get the best of both worlds. Low-pass sequencing:
Costs significantly less per sample than standard WGS.
Allows for richer, whole genome information compared with microarrays.
How can low-pass be accurate with such low coverage?
With the balance of information and cost in place, the question then becomes how is it possible to accurately genotype based on low-pass information? That’s where imputation comes in. In statistics, the basic definition of imputation is assigning values to missing data based on associated known data, and applying it to genetic data is no different. The goal of imputation in the setting of low-pass sequencing is to assign a genotype to an individual without direct measurements at every locus based on the statistical makeup of the genetic variants within a population. With enough representation of a population, low-pass sequencing data has been shown to be very accurate at assigning genotypes, as shown in cattle and for pharmacogenetics in humans, as well as increasing power for GWAS studies in dogs.
So why isn’t everyone doing low-pass sequencing and imputation for genotyping and GWAS?
The critical components of low-pass as a solution are 1) access to sequencing, 2) an understanding of the genetic variants in your population, and 3) statistical tools to impute genotypes based on the population knowledge. While generating sequencing data is becoming easier and less expensive over time, the other two take a bit of specialized knowledge. Building a reference panel for a population of interest and using mostly command-line tools to leverage that panel for imputation may not be skills readily available to researchers considering implementing low-pass.
That’s where Gencove comes in. As experts in genetics, low-pass sequencing, and imputation software, we provide an easy-to-use platform to house, analyze, and interpret low-pass sequencing data. Most projects can start right away on the platform, with reference panels for many species available now. For less common species or proprietary populations, tailored panels can be developed with the Gencove team of experts to ensure you’re maximizing the value of your data in the long term.
Once we’ve obtained your FASTQ files of low-pass sequencing data, you can access:
A suite of quality control measures to ensure confidence in your sequencing data
Genotypes for millions of genetic variants from your selected imputation reference panel
Sequence alignments against the reference genome in a portable format
Ancestry estimates from a set of 26 reference populations in human samples
Polygenic risk score estimates for CAD, breast cancer, and prostate cancer in human samples
Get the power of WGS at the cost of microarray
Pairing the continual technology developments making sequencing data more affordable with powerful platforms like Gencove to make low-pass data more easily interpretable, the future is bright for less biased, more comprehensive methods for genotyping.

Curious what the platform looks like and how to interact with it? Contact us for a free demo.