Sequencing projects come in many different forms. At Gencove, we’ve seen and supported a broad range of species and sample types. Occasionally, we receive projects that our partner service labs can't support using standard procedures, necessitating bespoke and innovative solutions.
One example is a recent project in which 392 Fast Technology for Analysis of nucleic acids (FTA) cards were used to collect canine saliva samples and the customer wanted to perform low-pass whole genome sequencing (lpWGS) on the DNA.
Although PCR approaches and amplicon sequencing have been performed, lpWGS of saliva on FTA cards has never been done before. The extremely low DNA yields made this a challenging sample type to work with.
Our service lab tried to extract DNA from the cards, but could not obtain enough DNA to perform our Gencove-modified, miniaturized KAPA HyperPlus library preparation (Roche). To ensure the projects completion, the FTA cards were sent to the Assay Development lab at the Gencove headquarters, where the team was confident we still had options for obtaining adequate DNA to perform lpWGS.
The lab team tested a number of elements of extraction protocols including:
The number of FTA card punches to use
The extraction buffer conditions
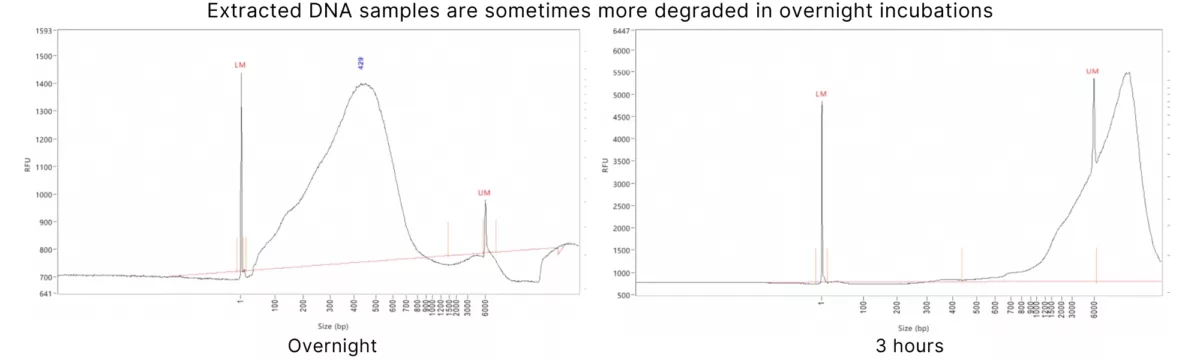
The extraction incubation times
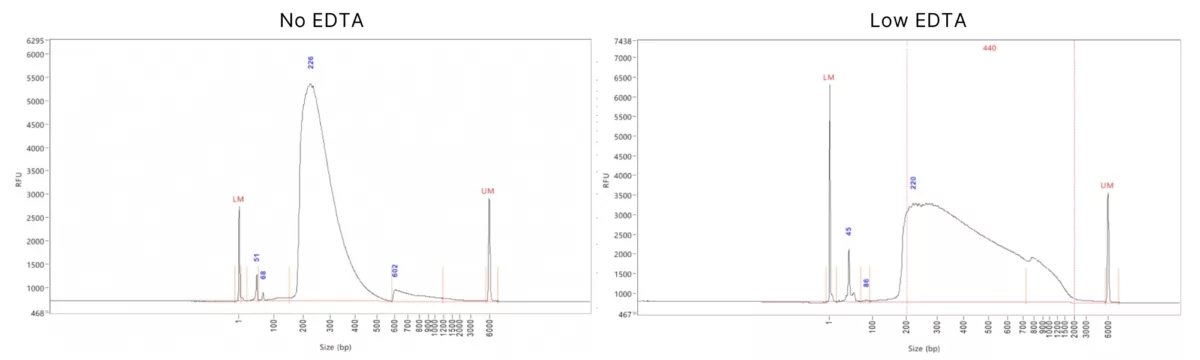
The elution buffer used for DNA extraction
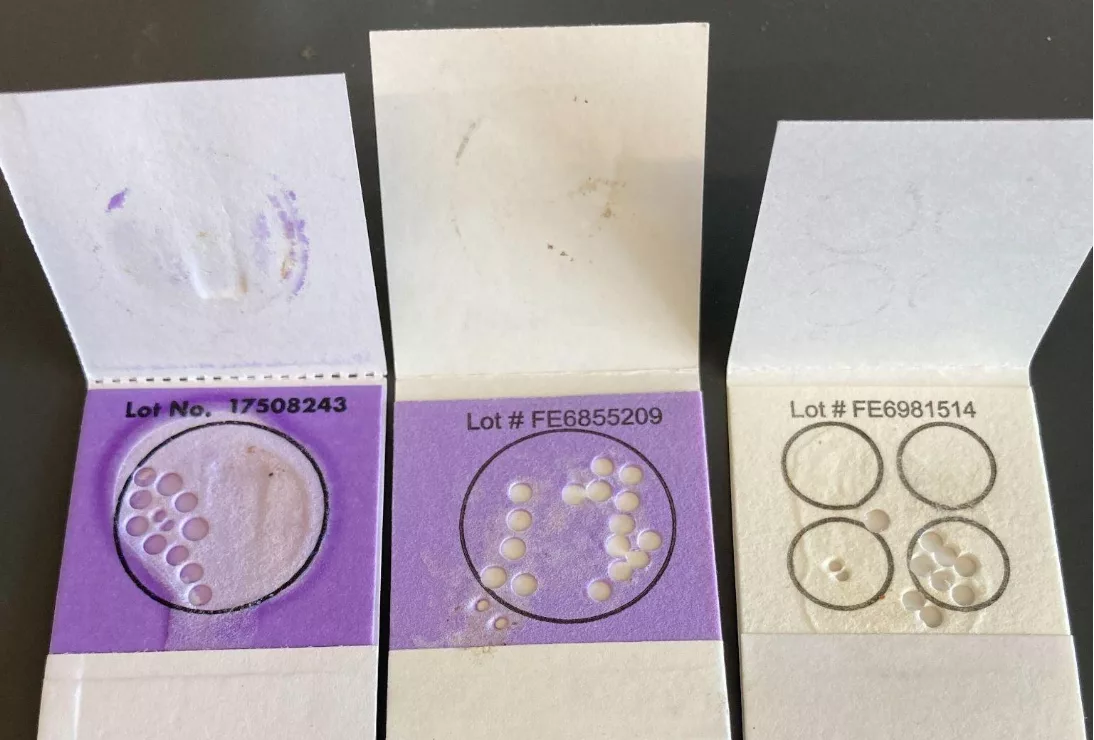
Punching the cards was not a trivial task. After some experimentation, we found that using 8 punches gave us better results than 4 punches. Punching 392 cards with 8 punches each required 3136 punches, which took a significant amount of time. Furthermore, FTA cards with indicators are normally purple and turn white where the sample is applied, however some cards had very little saliva applied which made it difficult to determine where to punch the cards (Figure 1). Additionally, some cards we received did not have any indicator on them at all, and it was hard to determine the region of the highest concentration of DNA.