Along with Warren Snelling and other colleagues at the USDA MARC (Meat Animal Research Center), we recently performed a study investigating the utility of imputation from low pass sequencing data in cattle in the context of genomic prediction for beef steers. This study is now published in MDPI Genes and can be accessed at the following link [0]. In this blog post, we briefly review the motivations behind this study and outline the main results.
Motivation
Commercial genotyping arrays are currently routinely used in genomic prediction for beef cattle due to the additional predictive validity that genomic data provides on top of pedigree information when modeling breeding values for phenotypes of commercial interest. Recent literature suggests that particularly for multi-breed populations, the inclusion of functional sequence variants can further increase the accuracy of such predictions [1,2,3]. However, one of the inherent limitations of currently available genotyping arrays for cattle is that they do not assay much of the known functional variation in beef cattle. Low pass sequencing (LPS) plus imputation to a haplotype reference panel addresses this limitation and streamlines the process, since one can accurately impute to a comprehensive set of functional variants, as the exact set of imputed variants can be tweaked at the software level without having to design a new assay at a molecular level.
Study Design
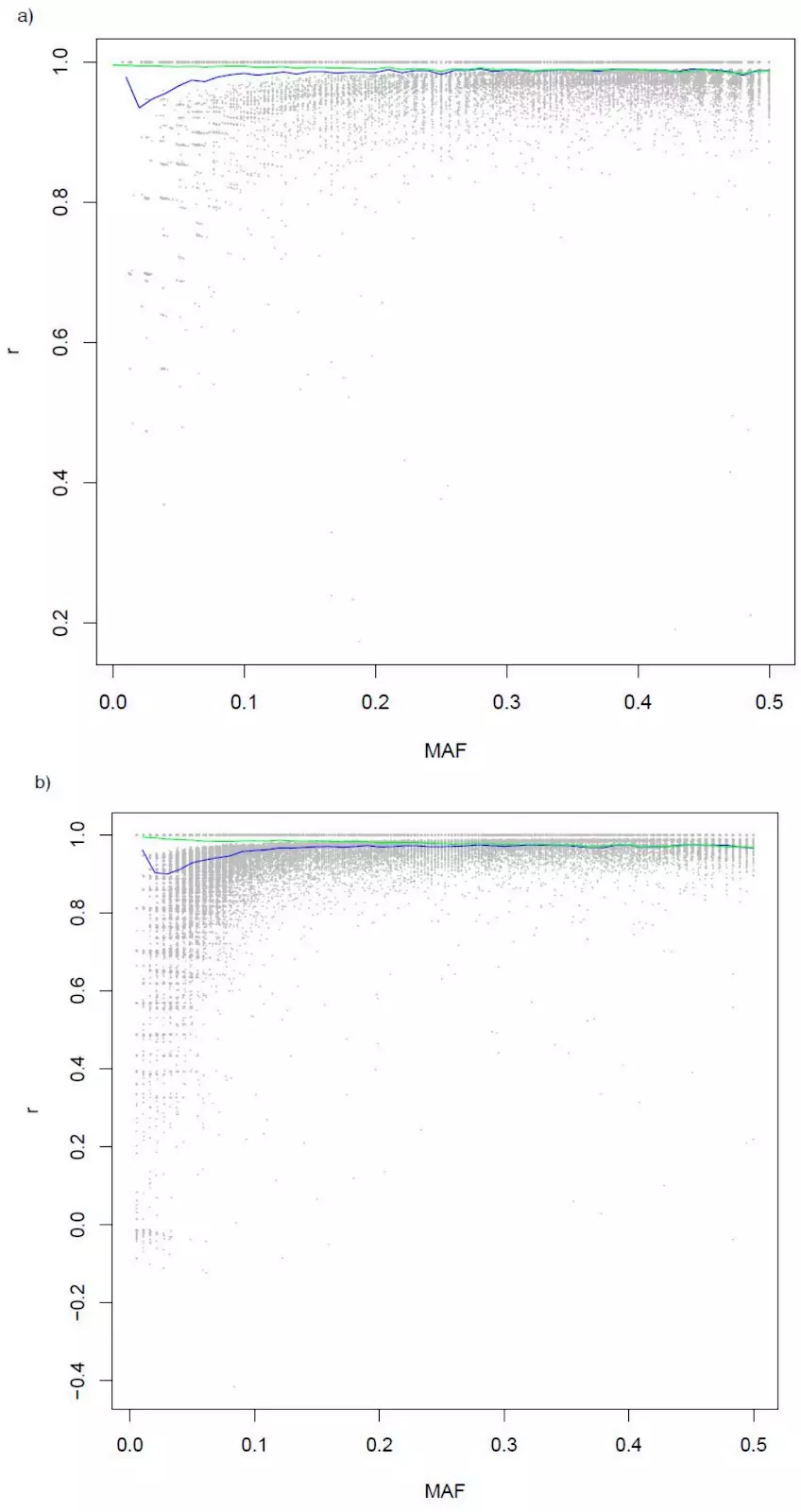
In this study, we evaluated the performance of low-pass sequencing in beef cattle by downsampling existing, high-depth (10x) sequence to the equivalent of 1x coverage for 77 steers, and imputed the data to a diverse haplotype reference panel comprising ~60MM variants and 946 individuals. These imputed genotypes were then compared to existing array data (genotype calls at a combined BovineHD + GGP-F250 variant set) for these same 77 individuals to examine the performance of imputation when compared to genotypes directly assayed at the molecular level (i.e., calls from a genotyping array).
> We then computed (1) estimated breeding values (EBVs) (models using the pedigree + known phenotypes), (2) genomic estimated breeding values (GEBVs) (models using genotypes + the pedigree + known phenotypes), and (3) estimated variant-level effect sizes, for three phenotypes of commercial interest: birth weight (BW), postweaning gain (PWG), and marbling (MARB) —all with the 77 steers held out of the training set. In the last model one does not directly estimate the breeding value as in (1) and (2), but rather, one applies the pre-estimated variant weights to genotypes from an arbitrary individual’s genotypes to obtain the individual’s molecular breeding value (MBV).
Reference Panel
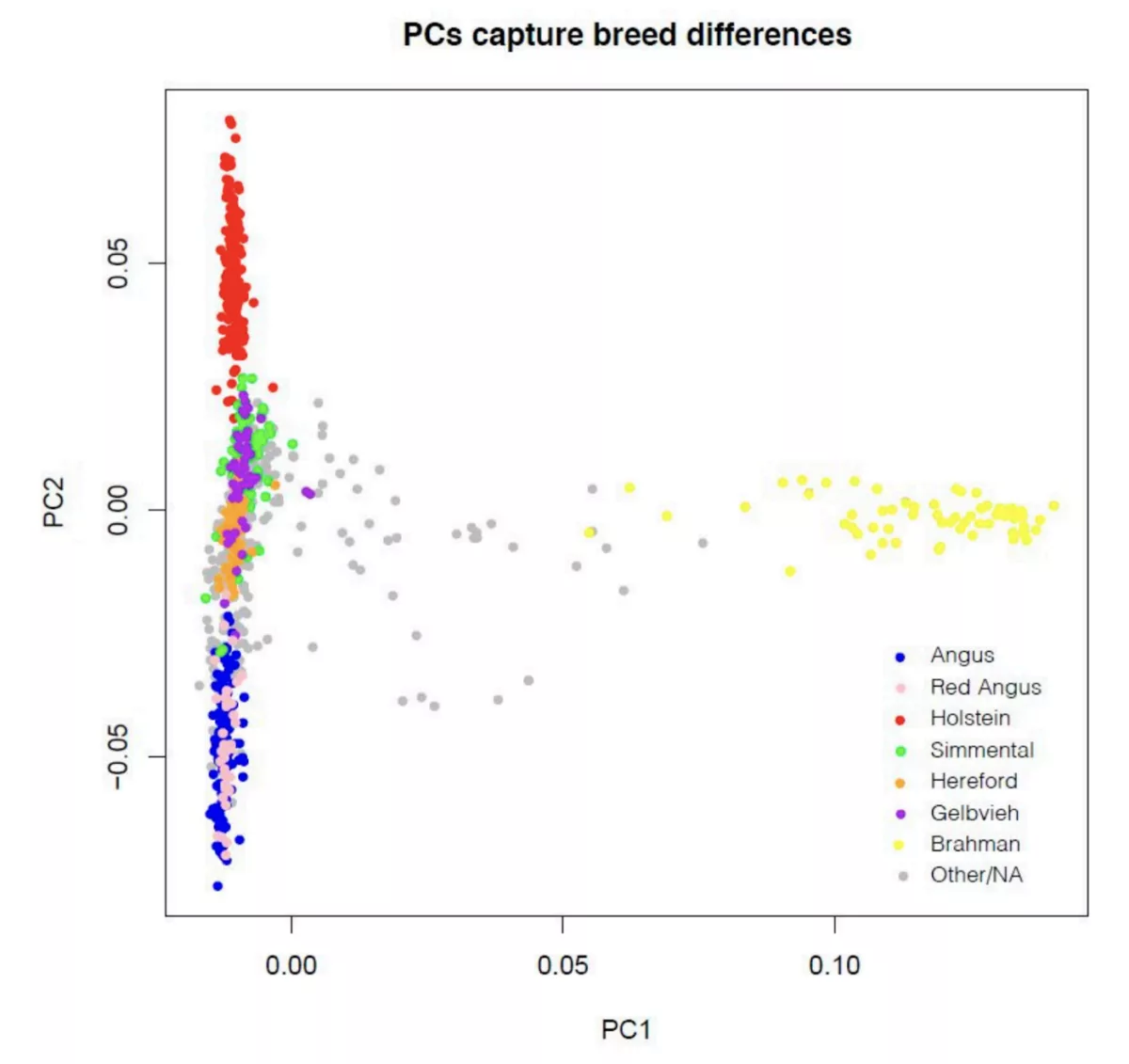
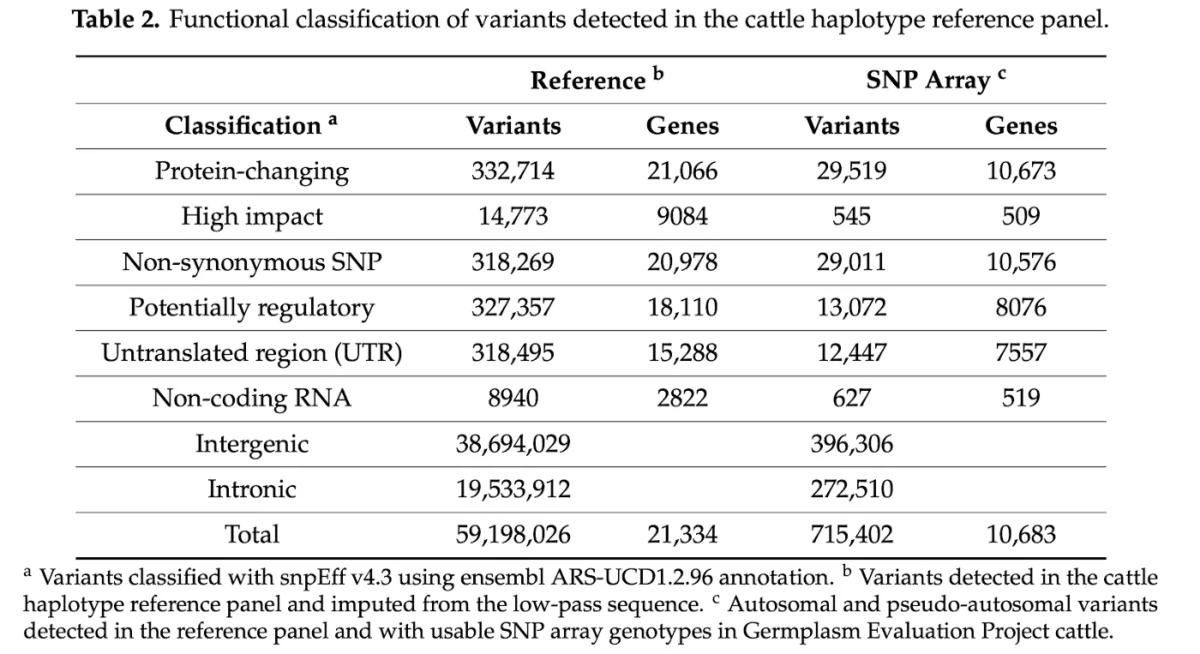
Before we describe the results of our study, we take some time to discuss the haplotype reference panel used. Over the last year and a half, we expanded our set of reference individuals from the ~500 individuals in our initial cattle product release (initially described here) to a set of 946 individuals spanning an even larger range of genetic diversity. At a high level, this reference panel comprises 946 individuals from B. taurus and B. indicus-related breeds, and spans the range of breeds most commonly found in the US (Figure 1). These genotype calls were generated using an in-house implementation of the GATK best practices pipeline and the haplotype reference panel comprises ~60MM SNPs and short indels.