Overview
At Gencove, our goal is to provide an end-to-end solution which facilitates reliable sample processing, sequencing, analysis, and data delivery for our partners. By leveraging the power of low-pass sequencing and imputation through a sophisticated technical stack, we process high volumes of samples at a reduced cost for our customers every day.
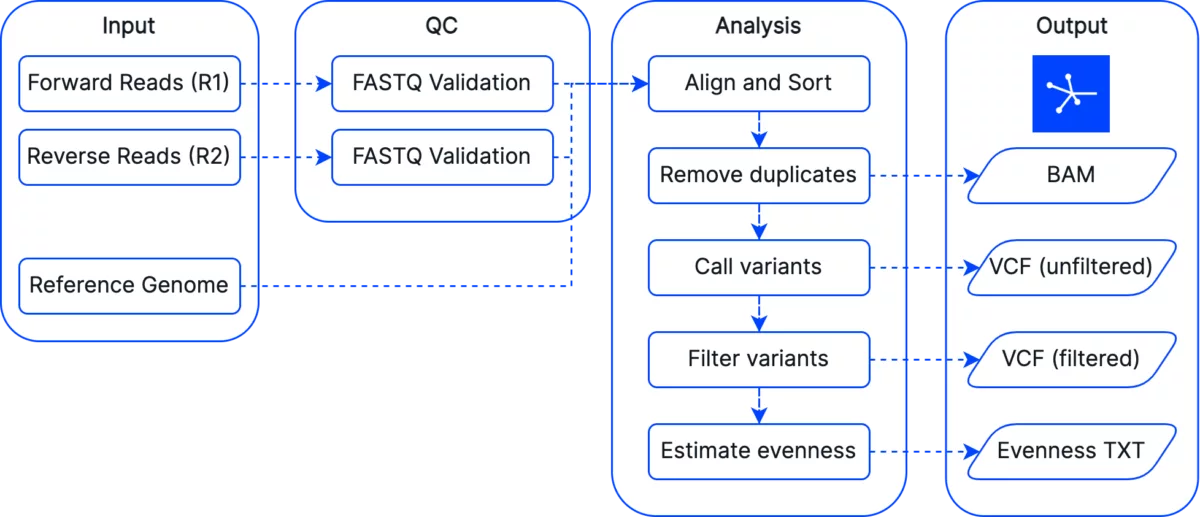
While low-pass sequencing followed by imputation is a powerful tool, use cases such as de novo discovery of variants, or detection of known rare variants, require sequencing at higher depths of coverage. To serve this use case and to help move towards Gencove’s vision of ubiquitous sequencing, we’re happy to announce support for our deep whole genome sequencing (WGS) pipeline!
Gencove can facilitate a cost effective approach to identifying rare variants in your samples through the WGS pipeline. We are happy to accommodate existing high coverage FASTQ datasets (e.g. >20X coverage), or to provide the lab support needed for an end-to-end process.
Usage
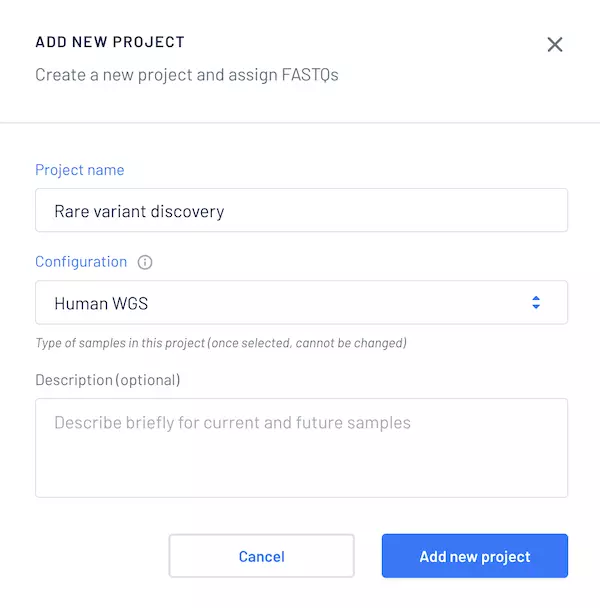
Our web platform, CLI, and API make it easy to work with your data. Running the pipeline is as simple as creating a project on the Gencove system with the relevant WGS configuration. The example below shows this configuration for humans; we are able to support other species as well.