RNA-sequencing has revolutionized human genomics by offering insights into gene expression and the broader transcriptomic landscape. This powerful technology is particularly impactful in fields such as oncology, drug development, and personalized medicine, where it allows researchers to identify biomarkers and develop targeted therapies by measuring gene expression levels and detecting RNA transcript mutations.
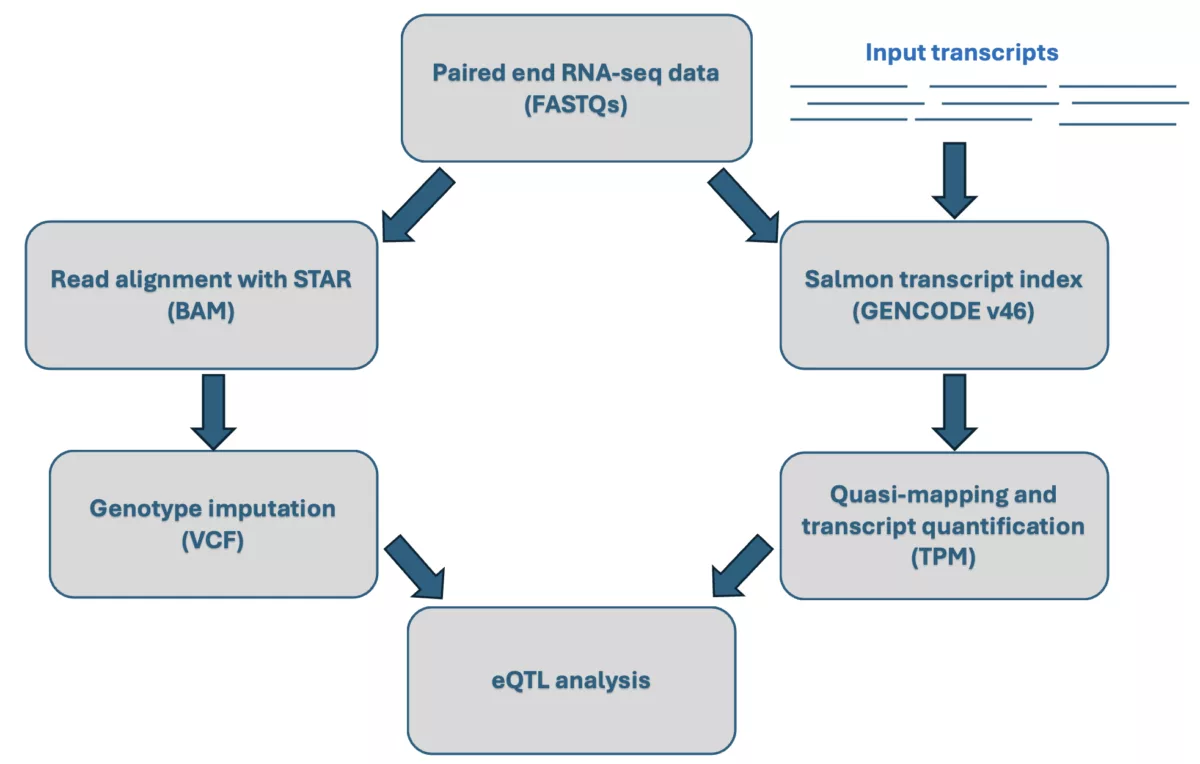
However, the potential of RNA-seq data can extend beyond expression analyses. One emerging use is leveraging complementary DNA (cDNA) reads from RNA-seq to perform genotype imputation1. While RNA-seq primarily captures reads from transcribed regions, these, when combined with off-target reads, provide a valuable backbone for genotype imputation. By leveraging these reads, we can impute missing genotypes to fill in the gaps in regions where there is limited or no sequencing coverage, and therefore complement the transcriptomic data with genome-wide genotypes without the need for additional sequencing. This dual benefit maximizes the value of RNA-seq data for more comprehensive and integrative downstream analyses such as eQTL studies that require genotype data.
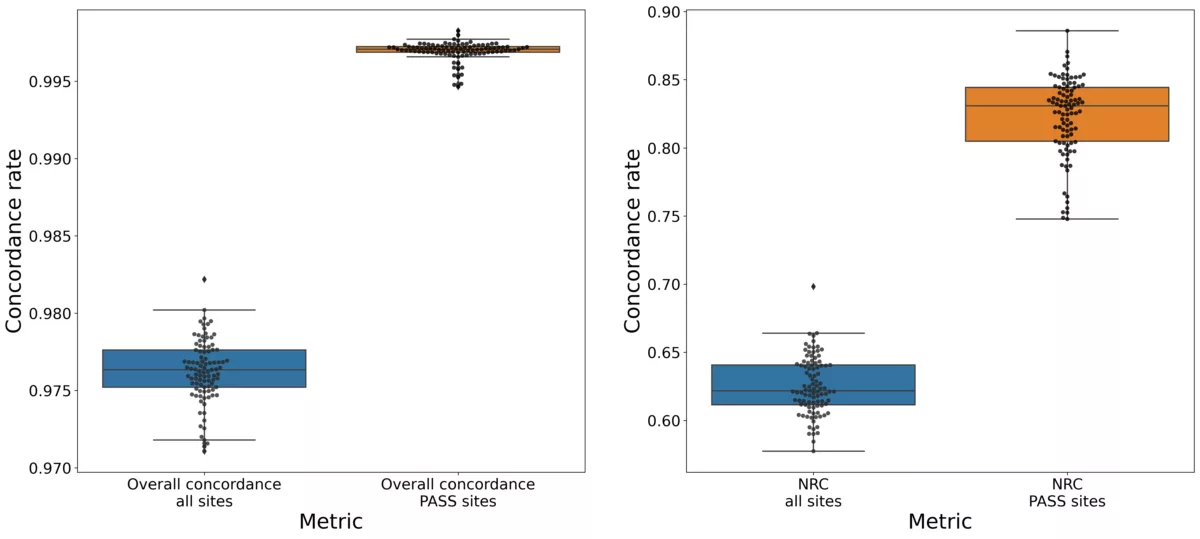
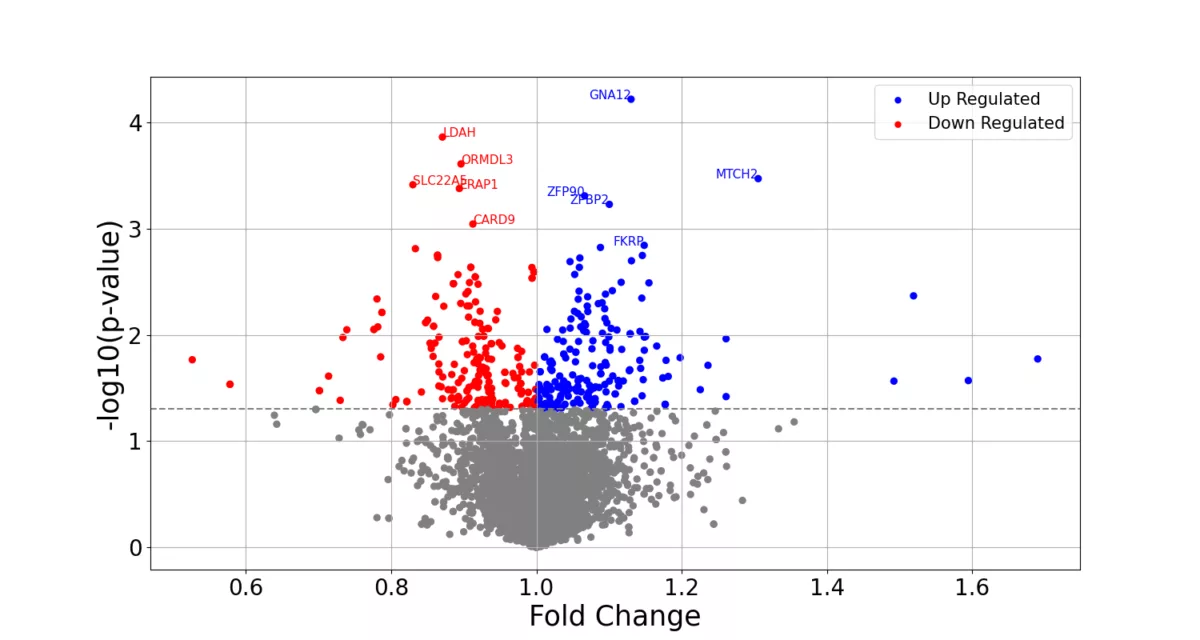
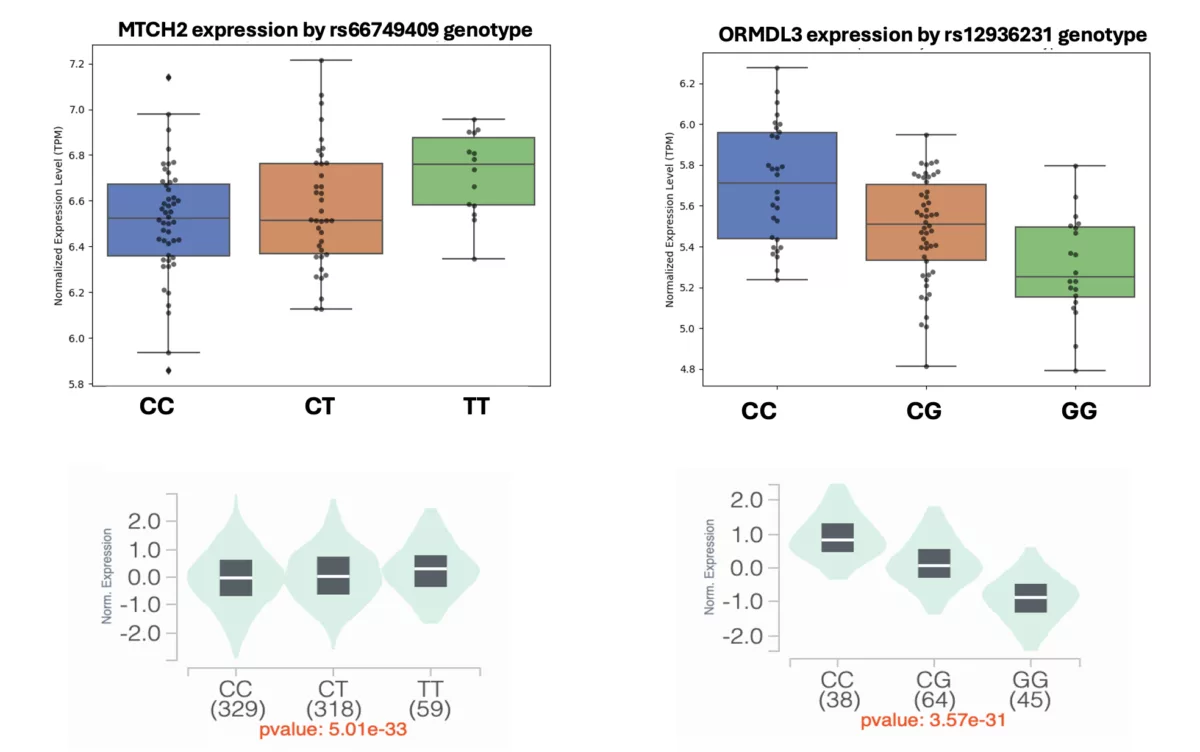
In this blog post, we outline an internal evaluation we performed to test the accuracy of genotype imputation from RNA-seq data, in addition to a subsequent replication of large effect-size cis-eQTL detection using the 1000 Genomes data.
Genotype imputation from RNA-seq Data
At Gencove, we offer a cost-effective solution to generate high quality genome-wide genotypes from low-coverage whole genome sequencing data through our advanced imputation pipelines. In this experiment, we used RNA-seq data from the 1000 Genomes Project Phase 1 2 to test the performance of our imputation pipeline with RNA-seq data as in input. This report outlines the results of this experiment, focusing on two main goals:
- Evaluating the accuracy of genotype imputation using RNA-seq data processed through the standard Gencove pipeline.
- Replicating large-effect cis-expression quantitative trait loci (cis-eQTLs) by integratively analyzing the imputed genotypes and gene expression data derived from RNA-seq.